




短碳链有机酸是生物质化学和生物转化的重要产物,但用途有限、附加值低,其高值化利用途径之一是通过酮基化反应同时实现碳链增长和脱氧[1]。短碳链有机酸酮基化是一个非常传统的反应,以丙酸为例,反应式如下:
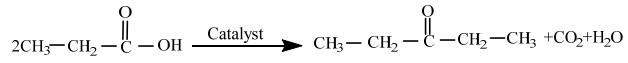
|
早在1858年Friedel等[2]报道了通过酮基化反应来制备丙酮。工业上,除了制备丙酮,异丁酸与乙酸高温交叉酮基化反应制备甲基异丙基酮也由德国康得阿维斯塔公司成功工业化[3]。目前世界上引领有机酸酮基化反应研究的是西班牙UPV-CSIC化学技术研究所的Corma教授课题组和美国University of Wisconsin-Madison的Dumesic教授课题组。Orozco等[4]深入开展了醛的催化酮基化反应研究;Serrano-Ruiz等[5]开发了一条从乙酰丙酸先生成γ-戊内酯、再至戊酸、接着酮基化和加氢制备长链烷烃的工艺。Pham等教授课题组[6]比较了有机酸碳链长度对催化酮基化反应的影响,结果表明碳链越长的有机酸越难发生酮基化反应。Lopez-Ruiz等[7]开展了通过LaxZryOz中金属组分的优化来提高酮基化催化剂的水热稳定性,在295℃下催化活性可以保持上百小时,但离工业化应用仍有距离。在国内,Wu等[8]研究了醋酸水相酮基化反应,发现ZrMnOx具有良好的醋酸酮基化催化活性;Ding等[9]研究了丙酸气相酮基化制备3-戊酮,结果表明,铈(Ce)-锆(Zr)复合氧化物具有较好的丙酸气相酮基化催化活性;Lu等[10]研究了CeO2中掺入Fe对有机酸气相酮基化反应的影响,发现少量Fe的掺入可显著提高有机酸酮基化反应活性。尽管有机酸酮基化反应近年来受到越来越多的关注,但研究多集中在高温、气相酮基化反应,反应温度通常为350~450 ℃。
近临界水一般是指温度范围为250~350 ℃的压缩液态水[11]。水在这一区域具有较大的电离常数因而自身具有一定的酸、碱催化能力,且具有能同时溶解有机物和无机物的特性。基于生物质基有机酸在许多情况下能与水共存的现状,本研究提出了在近临界水中将有机酸直接液相酮基化制备长链酮的思路。由于近临界水处于液相,且反应温度相对较低(< 350 ℃),因此,该方法具有能耗较低的优点。
实现近临界水中有机酸液相酮基化的关键是筛选出高效、水热稳定的催化剂。Gliński等[12-13]1995年至2014年连续发表了13篇文章,报道了有机酸金属氧化物催化酮基化反应,系统地比较了32种金属氧化物,发现Ce、Zr和Mn的氧化物具有高反应活性。受其启发,本研究拟以丙酸为模型物质,从Ce、Zr、Mn复合氧化物中筛选出近临界水中液相低温酮基化催化剂,以期为有机酸液相酮基化反应提供一种新思路。
2 实验部分 2.1 实验试剂与仪器 2.1.1 实验试剂硝酸铈六水合物(Ce(NO3)3·6H2O,质量分数为99.95%)、硝酸锆五水合物(Zr(NO3)4·5H2O,分析纯)、硝酸锰溶液(分析纯,质量分数为50%)、3-戊酮(质量分数为98%)均购自上海麦克林生化科技有限公司;硝酸镍六水合物(分析纯)、NaOH(分析纯)、无水Na2CO3(分析纯)、丙酸(分析纯)均购自国药集团化学试剂有限公司;磷酸(色谱纯)和乙腈(色谱纯)购自上海阿拉丁生化科技有限公司。
2.1.2 实验仪器1.67 mL微型高压反应釜,美国Swagelok公司;流化床沙浴炉,Techne SBL-2,英国Techne公司。
2.2 催化剂制备采用共沉淀法制备复合金属氧化物催化剂。以Ce-Zr金属氧化物为例介绍制备过程。
称取一定质量的Ce(NO3)3·6H2O和Zr(NO3)4·5H2O充分溶解于100 mL去离子水中,配成硝酸盐溶液;再称取一定质量的NaOH和无水Na2CO3充分溶解于200 mL去离子水中,配成混碱溶液。然后将硝酸盐溶液和混碱溶液同时缓慢滴入三口烧瓶中,控制pH值为9~10。充分沉淀后,70 ℃老化4 h,冷却至室温放置4 h。经过滤、去离子水洗涤三次后,置于110 ℃干燥箱中干燥12 h,最后在马弗炉中550 ℃下焙烧4 h制得CeZrOx催化剂。
2.3 催化剂活性评价先配制质量分数为5% 的丙酸水溶液,将0.8 mL丙酸水溶液(经计算在本研究最高反应温度340 ℃时反应釜内气相空间的占比超过20%)和催化剂加入微型高压反应釜中,旋紧反应釜保持密封。将流化床沙浴炉温度设置为反应所需温度,待升温至反应温度并稳定后,将密封好的反应釜放入流化床沙浴炉中反应一定时间。反应结束后,将反应釜取出置于冷水中急冷终止反应。冷却后,打开反应釜,将反应釜中液体转移到10 mL容量瓶中定容,经0.45 μm微孔水相滤膜过滤后,采用液相色谱(HPLC)对产物进行定量分析。丙酸转化率及3-戊酮收率按下式进行计算:
| $ 丙酸转化率\text{=}1-\frac{未反应的丙酸物质的量}{加入的丙酸物质的量}\times 100{\%} $ | (1) |
| $ 3\text{-}戊酮收率\text{=}\frac{实验实际所得3\text{-}戊酮的物质的量}{丙酸完全转化所得3\text{-}戊酮的理论物质的量}\times 100{\%} $ | (2) |
本研究每个实验点都进行了三次重复实验,数据为三次重复实验的平均值,误差是三次实验数据的标准偏差。
2.4 分析与表征反应物丙酸及反应产物3-戊酮采用HPLC分析(Agilent LC1260,美国Agilent公司),紫外检测、外标法定量。色谱柱为C18柱,流动相是质量分数为0.04% 的磷酸水溶液和乙腈,二者体积比为90:10,流速为1 mL·min-1,柱温为30 ℃,进样量为20 μL。对于丙酸,检测波长为210 nm;对于3-戊酮,检测波长为270 nm。
采用X射线衍射仪(XRD)、红外光谱仪(FT-IR)、透射电子显微镜(TEM)、比表面积及微介孔分析仪(BET)、X射线光电子能谱(XPS)、化学吸附仪(CO2-TPD)对部分Ce-Zr复合氧化物催化剂进行表征,表征仪器和条件如下:
XRD:XRD-6100,日本岛津公司,衍射源为波长0.154 nm的Cu kα,管电流为20 mA,管电压为40 kV,扫描衍射角范围为10°~90°;
FT-IR:Nicolet iS 10,美国Nicolet公司,采用KBr压片法进行制样;
TEM:JEOL JEM 2100F,日本JEOL公司;
BET:ASAP 2020,美国Micromeritics公司;
XPS:ESCALAB 250Xi,美国Thermo Fischer公司,工作电压为12 kV,灯丝电流为6 mA,通能为50 eV,步长为0.05 eV;
CO2-TPD:FINESORB-3010,浙江泛泰仪器有限公司,预处理温度为400 ℃,脱附温度为600 ℃。
3 实验结果与讨论 3.1 催化剂筛选 3.1.1 Ce-Zr-Mn金属氧化物的初筛采用正交实验法设计并合成了19种不同量比的Ce-Zr-Mn复合金属氧化物,在320 ℃下对其丙酸酮基化反应的催化活性进行了评价,结果如图 1所示,其他条件为反应时间10 h、丙酸量0.2 g、催化剂量0.1 g。由图可见,不同量比的Ce-Zr-Mn金属氧化物的酮基化反应活性存在显著差异,其中活性排在前三的分别是图中的A、B、C,即Ce2ZrOx、Ce2ZrMnOx和Ce2MnOx,它们的3-戊酮收率都接近40%。总的来看,单组分金属氧化物活性相对最差、三组分复合氧化物并没有表现出优于二组分,故本研究选择二组分复合氧化物为主要研究对象。虽然Ce-Zr金属氧化物和Ce-Mn金属氧化物酮基化反应活性相近,但考虑到Ce-Zr复合金属氧化物水热稳定性更具有优势,故以下选取Ce-Zr复合金属氧化物进行研究。

|
图 1 不同比例的Ce-Zr-Mn复合金属氧化物催化丙酸酮基化反应活性比较 Fig.1 Comparison of ketonization activity of propanoic acid over Ce-Zr-Mn metal oxides with different molar ratios |
在320 ℃下考察了不同量比的Ce-Zr复合金属氧化物对丙酸酮基化反应的影响,结果如图 2所示,其他条件为反应时间10 h、丙酸量0.2 g、催化剂量0.1 g。由图可见,Ce2ZrOx具有最佳的酮基化催化活性,其丙酸转化率和3-戊酮收率均高于其他比例的Ce-Zr氧化物。下面在催化剂表征的基础上,进一步开展Ce2ZrOx催化丙酸酮基化反应工艺和动力学方面的研究。

|
图 2 不同比例的Ce-Zr复合金属氧化物催化丙酸酮基化反应活性比较 Fig.2 Comparison of ketonization activity of propanoic acid over Ce-Zr metal oxides with different molar ratios |
为了探究复合金属催化剂活性的影响因素,对部分Ce-Zr复合金属氧化物开展了XRD、FT-IR、TEM、BET、XPS、CO2-TPD等表征。
3.2.1 XRD图 3为不同量比的Ce-Zr复合氧化物的XRD图谱,图中♣表示纯CeO2峰型、◆表示纯ZrO2峰型,虚线框内显示的是与Ce-O-Zr固溶体相关的CeO2(111)晶面的特征衍射峰变化。由图可见,CeO2的出峰位置为2θ=28.5°、33.1°、47.5°和56.3°(PDF#43~1 002),其对应的晶面分别为(111)、(200)、(220)与(311)。ZrO2出峰位置为2θ=30.4°、35.3°、50.7°、60.3°、63.3°以及74.8°(PDF#33~334)。由图还可见,纯ZrO2的半宽峰较窄,且峰形较为尖锐,而Ce-Zr复合氧化物的半宽峰较宽,且峰形较为平缓,这可能就是该催化剂酮基化活性较高的原因之一。随着Zr的摩尔分数增加,Ce-Zr氧化物的(111)晶面所对应的特征峰逐渐向高衍射角偏移,这说明一部分晶粒较大的Ce4+(0.097nm)被晶粒较小的Zr4+(0.084nm)所取代[9]。根据Scherrer公式[10],随着Zr的引入,Ce-Zr氧化物晶粒逐渐减小,同时这也说明Zr4+取代CeO2中一部分Ce4+,形成了Ce-O-Zr固溶体结构。

|
图 3 不同比例Ce-Zr复合氧化物的XRD图谱 Fig.3 XRD spectra of Ce-Zr oxides with different molar ratios |
图 4为不同Ce-Zr复合氧化物的红外图谱。其中3 400 cm-1处吸收峰为吸附水的─OH伸缩振动峰,1 600 cm-1处吸收峰为催化剂表面─OH官能团的弯曲振动峰[14]。纯CeO2具有较多种类的表面─OH基团,其中1 850、2 550 cm-1处的吸收峰分别对应Ce-OH的不同弯曲振动峰[15]。由图可见,纯ZrO2在1 850、2 550 cm-1处没有明显的振动峰,这说明纯ZrO2缺少表面─OH。740 cm-1处的振动峰为Ce-O结构的伸缩振动峰[16],并且随着Zr的引入,该振动峰逐渐向高波数偏移,且在880 cm-1处形成新的伸缩振动峰。根据文献[16]可推测880 cm-1处的振动峰为Ce-O-Zr结构上键联的─OH基团,这也进一步证明了Zr的引入导致了Ce-O-Zr固溶体结构的形成。

|
图 4 不同Ce-Zr复合氧化物的红外图谱 Fig.4 FT-IR spectra of different Ce-Zr oxides |
为探究催化剂颗粒的微观结构,对优选的Ce2ZrOx氧化物进行了TEM表征,结果如图 5所示,其中图 5(a)、(b)、(c)为不同放大倍数下复合氧化物的TEM图。由图可见,催化剂呈现均匀分散状态。图 5(d)为CeO2的衍射环,可见其是一个复杂的多晶结构。

|
图 5 Ce2ZrOx复合氧化物的TEM图 Fig.5 TEM image of Ce2ZrOx oxide |
开展了三种Ce-Zr氧化物的N2吸附、脱附等温测试,其比表面积、孔容和孔径结果如表 1所示,由表可见,Ce2ZrOx的比表面积SBET和孔容均显著大于CeO2和ZrO2。
|
|
表 1 不同Ce-Zr氧化物的比表面积、孔容和孔径数据 Table 1 Specific surface area, pore volume and pore size of different Ce-Zr oxides |
为探究不同量比的Ce-Zr复合金属氧化物表面化学价态及氧空位浓度,选取了4种Ce-Zr复合氧化物进行XPS表征,其Ce3d轨道的XPS图谱如图 6所示。根据XPS分峰拟合,Ce3d轨道可分为8个峰,其中u″′、u″、u′和u归属于Ce 3d3/2光电子能谱峰,v″′、v″、v′和v归属于Ce 3d5/2光电子能谱峰[17-18]。u′和v′归属于Ce3+,其余的为Ce4+光电子能谱峰[9]。Ce3+的含量为催化剂表面的氧空位浓度,Ce3+含量可通过u′和v′峰面积占整个Ce 3d轨道峰面积的比例计算,相关计算结果如表 2所示。由表 2可见,Ce-Zr复合金属氧化物表面Ce3+比例明显高于纯CeO2表面,说明Zr的引入提高了CeO2催化剂的氧空位浓度。其中Ce3+含量最高的催化剂为Ce2ZrOx,说明其具有最高的氧空位浓度,因而具有最佳的酮基化性能。

|
图 6 不同比例Ce-Zr复合氧化物的XPS图谱(Ce3d) Fig.6 XPS spectra of Ce-Zr oxides with different molar ratios(Ce3d) |
|
|
表 2 不同比例Ce-Zr复合氧化物的Ce3+含量 Table 2 Ce3+ content of different Ce-Zr oxides |
为了探究催化剂表面碱性位数量对酮基化反应的影响,开展了不同量比的Ce-Zr金属氧化物CO2-TPD表征,结果如图 7所示。将图中低于300 ℃的脱附峰归结为弱碱性位,而处于300~600 ℃的脱附峰归结为强碱性位。由图可见,纯CeO2在500 ℃左右具有较强的脱附峰,说明其强碱性位数量较多,而ZrO2在120 ℃左右具有较强的脱附峰,说明其弱碱性位数量较多[19]。随着Zr的引入,催化剂强碱性位数量逐渐减小,弱碱性位数量不断增多。由CO2-TPD曲线计算不同催化剂的脱附峰面积可得到其碱性位数量(强碱性位+弱碱性位)。计算结果如表 3所示。由表 3可见,Ce2ZrOx和CeZrOx催化剂碱性位数量高于纯CeO2和纯ZrO2催化剂,这可能与前者存在Ce-O-Zr固溶体结构有关[9]。其中Ce2ZrOx催化剂碱性位数量最多,进一步说明了其酮基化活性较佳的原因。

|
图 7 不同比例Ce-Zr复合氧化物CO2-TPD曲线 Fig.7 CO2-TPD curves of Ce-Zr oxides with different molar ratios |
|
|
表 3 不同Ce-Zr复合氧化物的碱性位数量 Table 3 Basic sites of different Ce-Zr oxides |
在320℃下开展了Ce2ZrOx催化剂用量对丙酸酮基化反应的影响研究,结果如图 8所示,其他条件还有反应时间为10 h、丙酸用量为40 mg。由图可见,对于不加任何催化剂的空白实验,只有非常少量的丙酸发生了转化,且无产物3-戊酮生成。当催化剂用量从10增加到40 mg时,丙酸转化率和3-戊酮收率均逐渐提高;继续增加催化剂量到50 mg时,丙酸转化率和3-戊酮收率无明显变化。催化剂用量为10~20 mg时,3-戊酮选择性一直保持90%以上;继续增大催化剂用量,3-戊酮选择性略有下降,原因可能是随着催化剂用量增大,生成了较多的副产物。因此,后续实验中催化剂用量选择20 mg。

|
图 8 Ce2ZrOx催化剂用量对丙酸酮基化反应的影响 Fig.8 Effect of Ce2ZrOx catalyst loading on ketonization of propanoic acid |
测定了310、320、330和340 ℃下Ce2ZrOx催化丙酸酮基化反应的动力学数据(其他条件为丙酸用量40 mg、Ce2ZrOx催化剂用量20 mg),结果如图 9所示,图 9(a)、(b)分别为不同温度下丙酸转化率、3-戊酮收率随时间的变化趋势。由图可见,随着反应温度升高,丙酸反应速率明显加快,当温度分别为310、320、330和340 ℃时,丙酸转化率达到60%所用时间分别为35、25、18和12 h。在反应温度为310 ℃下,经过50 h丙酸转化率才达到68%、产物3-戊酮收率才达到65%;而340 ℃时,只需30 h丙酸转化率可达85%、产物3-戊酮的收率可达81.6%。由图还可见,一定反应温度下,随着反应时间不断延长,丙酸转化率和3-戊酮收率的增长速度逐渐减慢,这是由于随着反应不断进行,底物丙酸的浓度逐渐减小。

|
图 9 反应温度和时间对丙酸酮基化反应的影响 Fig.9 Effects of reaction temperature and time on ketonization of propanoic acid |
丙酸酮基化反应生成3-戊酮的同时还有少量副产物生成,因此,该反应包括主副两个平行反应,如图 10所示。其中,A代表丙酸,B代表 3-戊酮,C代表其他副产物;k1为3-戊酮生成速率常数,h-1;k2为其他副产物生成速率常数,h-1。

|
图 10 丙酸酮基化反应 Fig.10 Ketonization of propanoic acid |
假设丙酸转化反应为一级反应,则其动力学方程为:
| $ \frac{{{\text{d}}{c_{\text{A}}}}}{{{\text{d}}t}}{\text{ = }} - k{c_{\text{A}}} $ | (3) |
式中:cA为丙酸摩尔浓度,mol·L-1;t为反应时间,h;k为丙酸反应速率常数,h-1。将上式积分可得:
| $ c_{\mathrm{A}}=c_{0} \mathrm{e}^{-k t} $ | (4) |
式中:c0为丙酸初始摩尔浓度,mol·L-1。
式(4)两边取对数可得-ln(cA/c0)=kt,将-ln(cA/c0)与反应时间t线性拟合,可得到不同反应温度下的k, 如图 11(a)和表 4所示,由表可见,随着反应温度升高,k逐渐增大。根据阿仑尼乌斯公式,将-lnk与1 000/(RT)线性拟合可得到丙酸转化反应活化能Ea,如图 11(b)所示,由图可见,Ea为98.9 kJ·mol-1。

|
图 11 丙酸酮基化反应活化能拟合 Fig.11 Activation energy fitting for ketonization of propanoic acid |
|
|
表 4 不同温度下的反应速率常数 Table 4 Rate constants at different temperatures |
假设丙酸反应生成3-戊酮及其他副产物均为一级反应,则其动力学方程可表示如下:
| $ \frac{\mathrm{d} c_{\mathrm{A}}}{\mathrm{d} t}=-\left(k_{1} c_{\mathrm{A}}+k_{2} c_{\mathrm{A}}\right) $ | (5) |
| $ \frac{{{\rm{d}}{c_{\rm{B}}}}}{{{\rm{d}}t}}{\rm{ = }}{k_1}{c_{\rm{B}}} $ | (6) |
式中:cB为产物3-戊酮摩尔浓度,mol·L-1。式(5)积分可得:
| $ {c_{\rm{A}}}{\rm{ = }}{c_{\rm{0}}}{{\rm{e}}^{ - \left( {{k_1} + {k_2}} \right)t}} $ | (7) |
式(7)可转化为:
| $ {\rm{ln}}\frac{{\rm{1}}}{{{\rm{1}} - {x_{\rm{A}}}}}{\rm{ = }}\left( {{k_1} + {k_2}} \right)t $ | (8) |
式中:xA为丙酸的转化率。将式(7)代入式(6)中,积分后得:
| $ {c_{\rm{B}}}{\rm{ = }}{c_{\rm{0}}}\frac{{{k_{\rm{1}}}}}{{{k_{\rm{1}}}{\rm{ + }}{k_{\rm{2}}}}}\left[ {1 - {{\rm{e}}^{ - \left( {{k_1} + {k_2}} \right)t}}} \right] $ | (9) |
将ln[1/(1-xA)]与反应时间t线性拟合,可得到不同反应温度下的(k1+k2)值,再将(k1+k2)值代入式(9),可得到不同温度下的k1和k2(见表 5)。分别将-lnk1、-lnk2与1 000/(RT)线性拟合可得到3-戊酮生成活化能Ea1为90.0 kJ·mol-1、其他副产物生成活化能Ea2为105.7 kJ·mol-1,可见,3-戊酮生成活化能低于副产物生成活化能,因而产物3-戊酮具有较高的选择性。
|
|
表 5 不同温度下的生成速率常数 Table 5 Formation rate constants at different temperatures |
在320 ℃下开展了Ce2ZrOx催化剂重复使用性能的测试,其他条件还有反应时间为10 h、催化剂用量20 mg、丙酸用量为40 mg。使用后的催化剂用10 mL去离子水洗三次,然后在干燥箱中110 ℃下干燥12 h,再重复使用,结果如图 12所示。由图可见,与新鲜催化剂相比,Ce2ZrOx催化剂重复使用三次后,3-戊酮收率和丙酸转化率并无明显下降,因此,Ce2ZrOx催化剂具有较好的水热稳定性。

|
图 12 Ce2ZrOx催化剂重复使用性能测试 Fig.12 Reuse performance of Ce2ZrOx catalyst |
(1) 筛选得到了近临界水中丙酸液相酮基化反应活性较佳的复合金属催化剂Ce2ZrOx,在340 ℃下反应30 h, 丙酸转化率为85%、3-戊酮收率为81.6%;
(2) Ce2ZrOx复合金属氧化物具有良好的重复使用性能;
(3) Ce2ZrOx具有较高酮基化催化活性和水热稳定性的原因是形成了Ce-O-Zr固溶体结构、较高的氧空位浓度、较大的比表面积和较多数量的碱性位;
(4) 丙酸液相酮基化反应活化能为98.9 kJ·mol-1,3-戊酮生成活化能为90.0 kJ·mol-1,其他副产物生成活化能为105.7 kJ·mol-1。
| [1] |
KUMAR R, ENJAMURI N, SHAH S, et al. Ketonization of oxygenated hydrocarbons on metal oxide based catalysts[J]. Catalysis Today, 2018, 302: 16-49. DOI:10.1016/j.cattod.2017.09.044 |
| [2] |
FRIEDEL C, UEBER S G. Gemischte acetone[J]. Justus Liebigs Annalen der Chemie, 1858, 108(1): 122-125. DOI:10.1002/jlac.18581080124 |
| [3] |
丁爽, 葛庆峰, 祝新利. 金属氧化物催化生物质衍生羧酸酮基化研究进展[J]. 化学学报, 2017, 75(5): 439-447. DING S, GE Q F, ZHU X L. Research progress in ketonization of biomass-derived carboxylic acids over metal oxides[J]. Acta Chimica Sinica, 2017, 75(5): 439-447. |
| [4] |
OROZCO L M, RENZ M, CORMA A. Cerium oxide as a catalyst for the ketonization of aldehydes: mechanistic insights and a convenient way to alkanes without the consumption of external hydrogen[J]. Green Chemistry, 2017, 19(6): 1555-1569. DOI:10.1039/C6GC03511F |
| [5] |
SERRANO-RUIZ J C, DUMESIC J A. Catalytic routes for the conversion of biomass into liquid hydrocarbon transportation fuels[J]. Energy & Environmental Science, 2011, 4(1): 83-99. |
| [6] |
PHAM T N, SHI D, RESASCO D E. Reaction kinetics and mechanism of ketonization of aliphatic carboxylic acids with different carbon chain lengths over Ru/TiO2 catalyst[J]. Journal of Catalysis, 2014, 314: 149-158. DOI:10.1016/j.jcat.2014.04.008 |
| [7] |
LOPEZ-RUIZ J A, COOPER A R, LI G S, et al. Enhanced hydrothermal stability and catalytic activity of LaxZryOz mixed oxides for the ketonization of acetic acid in the aqueous condensed phase[J]. ACS Catalysis, 2017, 7(10): 6400-6412. DOI:10.1021/acscatal.7b01071 |
| [8] |
WU K J, YANG M D, HU H S, et al. ZrMn oxides for aqueous-phase ketonization of acetic acid: Effect of crystal and porosity[J]. Chemistry-An Asian Journal, 2018, 13(9): 1180-1186. DOI:10.1002/asia.201800114 |
| [9] |
DING S, WANG H, HAN J Y, et al. Ketonization of propionic acid to 3-pentanone over CexZr1-xO2 catalysts: The importance of acid-base balance[J]. Industrial & Engineering Chemistry Research, 2018, 57(50): 17086-17096. |
| [10] |
LU F P, JIANG B B, WANG J D, et al. Insights into the improvement effect of Fe doping into the CeO2 catalyst for vapor phase ketonization of carboxylic acids[J]. Molecular Catalysis, 2018, 444: 22-33. DOI:10.1016/j.mcat.2017.05.022 |
| [11] |
吕秀阳, 何龙, 郑赞胜, 等. 近临界水中的绿色化工过程[J]. 化工进展, 2003, 22(4): 477-481. LYU X Y, HE L, ZHENG Z S, et al. Green chemical processes in near critical water[J]. Chemical Industry and Engineering Progress, 2003, 22(4): 477-481. |
| [12] |
GLIŃSKI M, KIJENSKI J, JAKUBOWSKI A. Ketones from monocarboxylic acids: Catalytic ketonization over oxide systems[J]. Applied Catalysis A: General, 1995, 128(2): 209-217. DOI:10.1016/0926-860X(95)00082-8 |
| [13] |
GLIŃSKI M, ZALEWSKI G, BURNOE, et al. Catalytic ketonization over metal oxide catalysts. XIII. Comparative measurements of activity of oxides of 32 chemical elements in ketonization of propanoic acid[J]. Applied Catalysis A: General, 2017, 470: 278-284. |
| [14] |
CHEN L, HE B, HE S, et al. Fe-Ti oxide nano-adsorbent synthesized by co-precipitation for fluoride removal from drinking water and its adsorption mechanism[J]. Powder Technology, 2012, 227: 3-8. DOI:10.1016/j.powtec.2011.11.030 |
| [15] |
KAZUO N. Infrared and Raman spectra of inorganic and coordination compounds[M]. New York: John Wiley & Sons, 2006.
|
| [16] |
HASAN M A, ZAKI M I. Oxide-catalyzed conversion of acetic acid into acetone: An FTIR spectroscopic investigation[J]. Applied Catalysis, 2003, 243(1): 81-87. DOI:10.1016/S0926-860X(02)00539-2 |
| [17] |
NELSON A E, SCHULZ K H. Surface chemistry and microstructural analysis of CexZr1-xO2-y model catalyst surfaces[J]. Applied Surface Science, 2003, 210(3/4): 206-221. |
| [18] |
REDDY B M, KHAN A. Structural characterization of CeO2-MO2(M=Si4+, Ti4+and Zr4+) mixed oxides by Raman spectroscopy, X-ray photoelectron spectroscopy and other techniques[J]. Journal of Physical Chemistry, 2003, 107(41): 11475-11484. DOI:10.1021/jp0358376 |
| [19] |
SNELL R W, HAKIM S H, DUMESIC J A, et al. Catalysis with ceria nanocrystals: Bio-oil model compound ketonization[J]. Applied Catalysis A: General, 2013, 464: 288-295. |