




2. 浙江大学 能源清洁利用国家重点实验室, 浙江 杭州 310027;
3. 浙江大学 药学院, 浙江 杭州 310058
2. State Key Laboratory of Clean Energy Utilization, Zhejiang University, Hangzhou 310027, China;
3. College of Pharmaceutical Sciences, Zhejiang University, Hangzhou 310058, China
塑料的大量使用导致塑料垃圾的处置成为了一个亟待解决的全球性问题[1]。在众多的处置方法中,热解和气化等热处置方法在减少土地资源侵占和提高能源利用方面具有优势。一般来说,除了聚苯乙烯(polystyrene,PS)和聚对苯二甲酸乙二醇酯(polyethylene terephthalate,PET)生成芳香化合物,其余绝大部分废塑料经热裂解反应生成的初级产物都是聚烯烃等链烃类化合物[2]。其中,轻质系列链烃化合物(C2~C4)在焦油的形成过程中起着关键的作用,这类化合物可以进一步通过重整反应生成不凝性气体,也可以彼此结合通过加乙炔脱氢或者Diels-Alder反应,进一步环化和芳香化并生成芳香化合物。
蒸气是一种较为常见的气化剂,可以通过蒸气重整反应减少产物中焦油的含量,并进一步提高H2产率[2]。在水蒸气参与气化反应时,实质上是通过H2O分子产生的H/O/OH自由基与轻质链烃化合物进行反应的。已有学者利用激波管反应器及配套的检测设备对轻质烃类化合物与H[3-4]、O[5-6]、OH[7-8]的反应速率进行了研究。然而,由于链烃化合物的反应时间短[9],并且存在多种反应路径,试验的方式很难对单一的反应过程进行精准地检测和分析。
Gaussian软件已被广泛应用于有机化合物的精确计算[10-13],并且许多研究人员通过与相关试验结果的比较验证了计算的准确性。目前,也有许多关于链烃化合物计算的文献报道,尤其是对于C2系列链烃化合物的反应机理已进行了较为深入的研究。然而,针对C3系列链烃化合物的研究虽然有报道,但是大多仅仅是针对单一的链烃化合物或者单个自由基参与的反应过程进行研究。比如,丙烯或丙炔与O自由基[14-16]、H自由基[10, 17]、OH自由基[11]的反应。本研究采用Gaussian及其配套软件从微观的角度分析了丙烷、丙烯和丙炔的自身分解或者是在H2O、O/H/OH/CH3自由基作用下的裂解反应,完善了典型C3系列链烃化合物的裂解机理。
2 计算方法本研究主要使用了Gaussian 16[18]、辅助视图软件(GaussView 6.0)以及Shermo软件[19]对C3系列链烃化合物的反应过程进行模拟计算。
首先,进行密度泛函理论(density functional theory,DFT)方法B3LYP/6-31G(d)基组下的过渡态计算,并对得到的过渡态结果进行虚频的唯一性验证以及内禀反应坐标(IRC)计算以确保过渡态猜测的合理性。然后,在B3LYP/6-311G++(d, p)下计算所有化合物的自由能,并结合相应的能垒判断整个反应过程的决速步。在此过程中,所有关于B3LYP泛函的计算均添加了色散校正。为了进一步得到具有更高精度的计算结果,耦合簇理论的应用是必不可少的[20-21],但是完整的耦合簇方法计算量太大,在实际计算过程中大多做了相应的简化[22-23]。目前比较常用的是非迭代三重激发态的耦合簇CCSD (T)与Dunning相关一致基组[24]相结合的计算组合模式。本研究在对反应过程中的反应物、过渡态及产物进行了相应的优化和振动分析后,采用CCSD(T)/cc-pVTZ对这些化合物进行了更高精度下的单点能计算。随后,将计算结果导入Shermo软件进行热物性特性参数的计算。在此过程中,零点能量(ZPE)采用的校正因子为0.988 7[25]。具体的计算流程如图 1所示。

|
图 1 模拟计算过程 Fig.1 Simulation calculation process |
由Shermo计算得到的各个化合物的热物性参数可以代入式(1) 计算反应速率常数。相应的计算公式如下:
| $ K\text{(}T\text{) = }\frac{\kappa \text{(}T\text{)}\sigma {{k}_{\text{B}}}T}{h}{{\text{(}\frac{RT}{{{p}_{\text{0}}}}\text{)}}^{\Delta n}}\text{exp}\ [\frac{-\Delta G\text{(}T, {{s}_{\text{0}}}\text{)}}{{{k}_{\text{B}}}T}]$ | (1) |
式中:κ(T)为基于Skodje-Truhlar方法计算得到的量子力学隧道效应的透射系数[26];𝜎为反应对称系数;Δn=n−1(n为反应分子数);T为热力学温度,K;p0为标准大气压,p0= 105 Pa;ΔG(T,s0)是根据反应决速步理论[27]确定的过渡态与反应物之间的自由能垒;s0为决速步所对应过渡态在反应坐标系上的位置;R、kB和h分别为理想气体常数、玻尔兹曼常数和普朗克常数。
3 结果与讨论 3.1 C3系列链烃化合物的反应路径及过渡态烷烃的热分解是碳基燃料燃烧化学中的重要过程,除了其自身的裂解外,还可以通过夺取其上的原子或自由基来破坏原有分子结构[28]。在讨论丙烷裂解生成C2化合物的可能反应路径时,不仅考虑了丙烷在高温条件下的热裂解反应,还考虑了丙烷在水蒸气和H/OH/CH3自由基作用下的气化反应,具体反应如下:
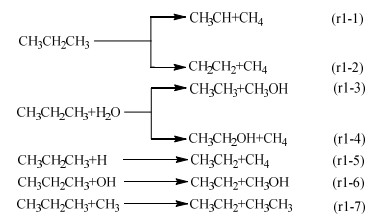
|
已有研究人员针对丙烷分解为甲基和乙基的裂解路径进行了研究[28-31],在此基础上进一步探索了丙烷通过氢转移实现自身裂解的反应机理,2种反应过程分别如路径r1-1和r1-2所示。结合过渡态结构来看,丙烷的热裂解可以通过中间碳原子上的氢转移到链端甲基碳上(路径r1-1),或者是一端甲基上的氢转移到另一端甲基上(路径r1-2)来促使C─C键断裂。当水蒸气和丙烷反应时,由水分子生成的OH自由基可分别与丙烷的链端碳原子或中间碳原子结合,生成的H自由基则是与剩下的基团结合,如路径r1-3和r1-4所示。此外,H/OH自由基以及反应过程中生成的甲基都能促进丙烷的分解,如路径r1-5、r1-6和r1-7所示。鉴于通过H/OH自由基进行简单的夺氢反应来改变原有结构的反应路径众多,但是并没有实现真正意义上的脱碳,这里将不再进一步展开讨论。
丙烯是一种重要的石化原料,也是C4及以上的烷烃在热裂解或者氧化过程中形成的重要中间体[32]。与丙烯有关的分解反应如下:
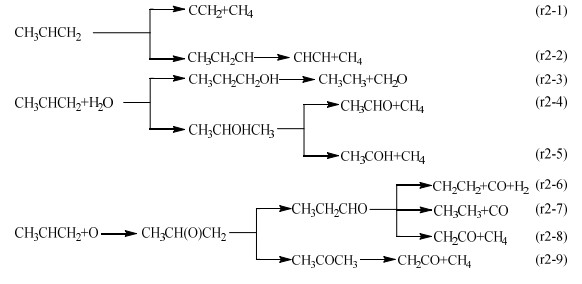
|
|
丙烯的自身裂解可以通过中间碳原子上的氢转移至链端的甲基碳上(路径r2-1)来实现。同时,当丙烯中的甲基从双键的一端转移至另一端后,也可以进一步通过氢转移(路径r2-2)来实现C─C键断裂。从计算结果来看,丙烯的裂解过程并非是双键上氢的转移,而是链端甲基整体迁移的结果,这与Hung等[32]的计算结果一致。当水蒸气和丙烯反应时,首先进行的是加成反应。随后,生成的中间产物正丙醇(CH3CH2CH2OH)或异丙醇(CH3CHOHCH3)可以通过羟基氢向其相邻碳上的转移实现进一步的裂解,如路径r2-3、r2-4所示。同时,异丙醇也可以直接由中间碳原子上的氢转移实现C─C键断裂(路径r2-5)。
此外,丙烯上的不饱和双键也可以与O/H/OH自由基进行加成,并且在经历一系列的旋转和结构重排后实现中间产物的进一步裂解。比如O自由基不仅可以通过与双键末端或者中间碳原子成键[16]的方式来实现丙烯加成,也可以与丙炔进行类似的加成反应[14]。但是从这些文献展示的能垒图中可以发现,加成反应是容易实现的,其所对应的能隙都很小;也有研究认为O自由基与不饱和链烃上的碳碳双键或三键结合是一个自发过程,没有对应的过渡态[33-36]。由于本研究在过渡态计算过程中使用的基组精度有限,无法根据文献[14, 16]中提供的分子结构验证相应的结果,也未能找到其他合适的过渡态结构,因此本研究将相应的加成看作一个极易自发进行的过程,后续的讨论将围绕加成之后的反应展开。
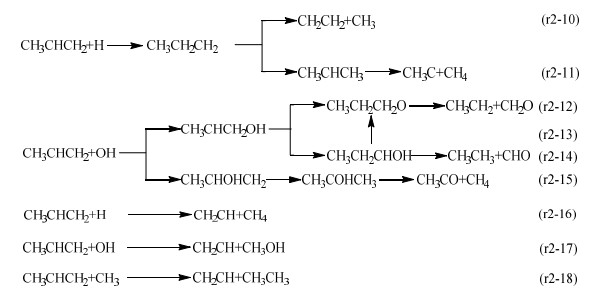
丙烯和O自由基通过加成反应生成CH3CH(O)CH2后,还会进行氢转移反应。由于氧原子有2个加成位点,因此产物分别为丙醛(CH3CH2CHO)和丙酮(CH3COCH3)。其中,丙醛的进一步裂解可以由醛基上的氢与链端甲基上的氢结合(路径r2-6)或者转移到中间碳原子上(路径r2-7)来实现。同时,丙醛中间碳原子上的氢转移至链端甲基并带动醛基上的氢迁移(路径r2-8)也能够促使C−C键断裂。丙酮则主要是通过一端甲基上氢原子转移到另一端甲基碳上的方式来裂解并生成CH2CO和CH4(路径r2-9)。同样的,H和OH自由基也可以与丙烯进行加成反应。H自由基与丙烯结合生成的CH3CH2CH2可以直接脱甲基生成CH2CH2和CH3(路径r2-10),也可以在经历2次氢转移后生成CH3C和CH4(路径r2-11)。由于OH自由基与双键碳的结合位置不同,可以生成2种中间体(CH3CHCH2OH或CH3CHOHCH2)。其中,CH3CHCH2OH可以由OH基上的氢经不同的方式迁移至中间碳原子,继而脱除异构体上的CH2O形成CH3CH2,如路径r2-12、r2-13所示。同样的,与链端碳原子相连的氢原子直接转移至中心碳也可以促使CH3CHCH2OH转化成CH3CH2CHOH,而后裂解生成CH3CH3和CHO(路径r2-14)。CH3CHOHCH2则是由中心碳上的氢转移至链端的亚甲基上形成CH3COHCH3,然后进一步裂解生成CH3CO和CH4(路径r2-15)。此外,H自由基(路径r2-16)、OH自由基(路径r2-17)、甲基自由基(路径r2-18)还可以通过夺取丙烯链端的甲基促使丙烯分解。
具有不饱和键的丙炔不仅是化石燃料在热反应过程中重要的中间产物,也是影响多环芳烃或者炭黑形成的关键因素[5, 14, 37]。有所不同的是,丙炔在热处置过程中很难通过氢转移等方式直接实现热裂解。Miller等[38]计算了多种有关丙炔的反应路径,但是这些反应仅能够将丙炔转变成异构体或者以释放H2的形式进行脱氢反应,而无法将丙炔转化成C2及以下的化合物。此时,H2O分子和自由基对于促进丙炔的裂解显得尤为重要,具体反应如下所示:
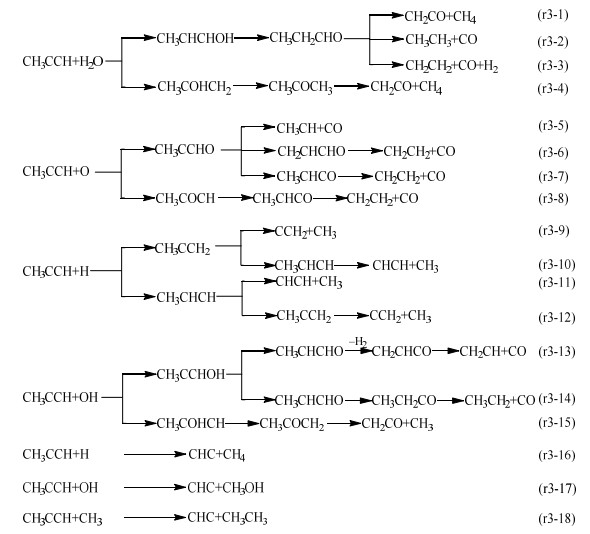
|
当水蒸气参与丙炔的气化反应时,首先会以加成的方式生成CH3CHCHOH或者CH3COHCH2,后续的裂解反应则是与丙烯和O自由基加成后的转化路径类似,都是通过氢转移的方式促使C─C键断裂,具体的反应流程如路径r3-1到r3-4所示。当丙炔和O自由基反应时,也有2种加成方式分别生成CH3CCHO和CH3COCH。当加成产物为CH3CCHO时,醛基上的氢转移至中心碳原子,可以促使C─C键断裂(路径r3-5);同时,如路径r3-6和r3-7所示,醛基上的氢和链端甲基上的氢转移至中心碳原子后,也可以促使中间产物进一步裂解生成CH2CH2和CO。当加成产物为CH3COCH时,相应的裂解过程则主要通过甲基转移来实现(路径r3-8)。
类似的,H和OH自由基都可以与三键碳原子结合,并且根据成键位置的不同可以形成2种加成产物。路径r3-9和r3-11分别展示了H自由基与丙炔进行加成反应后,中间产物(CH3CCH2或CH3CHCH)进一步脱除链端甲基的反应过程。此外,2种加成产物还可以通过氢转移彼此转换,随后脱除链端的甲基,甲基形成C2及以下的化合物,相应的反应过程如路径r3-10和r3-12所示。当OH自由基与丙炔进行加成反应时,加成产物可以通过OH基上的氢转移形成更加稳定的CH3CHCHO或CH3COCH2。其中,CH3CHCHO可以由甲基和醛基上的氢彼此结合释放H2,继而脱除CO(路径r3-13);也可以通过氢转移形成CH3CH2CO后再进一步脱除CO(路径r3-14)。CH3COCH2则是可以直接脱除链端的甲基生成CH2CO(路径r3-15)。最后还考虑了H自由基(路径r3-16)、OH自由基(路径r3-17)、CH3自由基(路径r3-18)与丙炔链端甲基的反应。
3.2 C3系列链烃化合物的反应动力学参数图 2和3分别展示了丙烷在不同条件下的反应能垒E和速率常数K,从图中可以看出,丙烷的自身裂解速率受温度的影响非常大,当温度从600升至1 000 ℃,路径r1-1和r1-2的反应速率常数分别有8个和9个数量级的提升。其中,路径r1-1是更容易进行的,相应的速率常数要比相同温度下路径r1-2的速率常数高出3~4个数量级,这也说明丙烷的裂解更容易通过中间碳原子上的氢转移至链端的甲基来实现。但是相较于文献中的结果,由于反应路径和裂解产物的不同,路径r1-1在同等温度下的速率常数要低4~6个数量级[28-29]。

|
图 2 丙烷的能垒 Fig.2 Energy barriers of propane |

|
图 3 丙烷在不同反应路径下的速率常数,其中r1-1, r1-2为1级反应(s−1),其余均为2级反应(L·mol−1·s−1) Fig.3 Reaction rate constants of propane under different reaction paths. r1-1 and r1-2 are first-order reactions (s−1) and others are second-order reactions(L·mol−1·s−1) |
类似的,温度的升高对于促进水蒸气和丙烷的反应也是十分有利的。当温度升至1 000 ℃时,路径r1-3和r1-4的反应速率常数均提高了9个数量级,路径r1-4的速率常数比相同温度下路径r1-3的速率常数略大,但没有出现数量级的差异。同时,相较于水蒸气直接作用于丙烷的裂解,H自由基、OH自由基和甲基都能够很好地促进丙烷的裂解,相应的反应路径从易到难分别是r1-5、r1-6、r1-7。其中,甲基与丙烷的反应速率常数相对较低(路径r1-7),但仍比相同温度下水蒸气与丙烷反应(路径r1-3)的速率常数高出11~14个数量级,这可能是因为水蒸气与丙烷的反应还涉及H2O的裂解过程,因此需要提供更多的能量才能实现这一过程。由于目前鲜有报道自由基夺取甲基的反应,这里仅能参考H/OH/CH3自由基夺取链端氢原子的反应[39-41],相应的速率常数要比本研究中路径r1-5到r1-7的速率常数高出至少5个数量级。这意味着丙烷在自由基的作用下断开碳链是可行的,但是更容易进行的是H/OH/CH3自由基夺取链端氢原子的反应。
图 4展示了丙烯在不同条件下的反应能垒。其中,路径r2-1、r2-16到r2-18属于单步反应,其余均为多步反应。根据决速步理论[27]可以进一步确认路径r2-11、r2-14和r2-15的决速步是第3步,而其余多步反应的决速步均为第2步。

|
图 4 丙烯的能垒 Fig.4 Energy barriers of propylene |
当水蒸气和丙烯进行反应时,3种反应路径从易到难的顺序是:路径r2-5、r2-4、r2-3。结合3.1节的介绍可知,3种反应路径的关键步骤是氢转移促使C─C键断裂的过程(第2步)。其中,路径r2-3和r2-4都涉及羟基脱氢的过程,相应的速率常数比相同温度下路径r2-5的速率常数低1~3个数量级,这说明在此反应条件下O─H键的断裂比C─H键的断裂更加难以进行。相较于丙烷而言,丙烯在水蒸气作用下的裂解容易得多。在相同温度条件下,丙烷和H2O分子在最优反应路径下的速率常数(r1-4)比路径r2-3的速率常数低3~6个数量级。这表明H2O分子更易在不饱和键处加成,然后通过氢转移促进中间产物的裂解。
如图 5所示,在O自由基与丙烯的4条反应路径中,路径r2-6的反应速率常数是最大的。随着温度从600增加到1 000 ℃,路径r2-6的反应速率常数从3.290×10−2增至3.824×104(L·mol−1·s−1),比另外3个反应路径在相同温度下的速率常数要高出至少2~3个数量级。这是因为O自由基与双键端碳结合生成的丙醛比与中间碳原子结合生成的丙酮活性更高,因此相同条件下丙醛更容易裂解并生成H2和CO。图 5中路径r2-10是所有路径中最容易进行的,这充分表明H自由基对促进丙烯的裂解十分关键。H自由基与丙烯双键加成生成CH3CH2CH2后,可以有效促使链端甲基的脱除(路径r2-10),相应的速率常数与文献[4, 42]报道的结果一致,并且比相同温度下路径r2-11的速率常数高出11~15个数量级。结合路径r2-11的能垒图,当CH3CH2CH2通过氢转移形成更加稳定的CH3CHCH3自由基后,若要进一步通过氢转移裂解成CH3CH和CH4,所需的能量将大大增加,路径r2-11的速率常数也会明显减小。

|
图 5 丙烯在不同反应路径下的反应速率常数随温度的变化情况,其中路径r2-1和r2-2为1级反应(s−1),其余均为2级反应(L·mol−1·s−1) Fig.5 Reaction rate constants of propylene under different reaction paths. r2-1 and r2-2 are first-order reactions (s−1) and others are second-order reactions (L·mol−1·s−1) |
当OH自由基参与丙烯的裂解反应时,4种可能的反应路径都涉及氢转移过程,其中最容易进行的是路径r2-12,相应的速率常速从600 ℃的3.806×105增至1 000 ℃的1.181×108(L·mol−1·s−1)。这说明OH自由基与丙烯结合更容易促使羟基中的氢原子转移至中间碳原子上,从而断开C─C键。路径r2-13则是经过连续的2次氢转移来实现加成产物的进一步分解,相应的速率常数虽然比相同温度下路径r2-12低2个数量级,但是仍比路径r2-15高出8个数量级。这主要是因为OH自由基与中间碳原子结合,随后通过氢转移形成了相对更稳定的CH3COHCH3,从而使得路径r2-15后续的裂解反应变得更加困难。路径r2-14则是4个反应中最难发生的。虽然一开始生成了同路径r2-12和r2-13一样的加成产物,但是在氢转移形成CH3CH2CHOH后,则是直接将羟基上的氢原子转移至中间碳原子上, 实现C─C键的断裂。这表明羟基中的氢原子转移过程也是促使中间产物进一步裂解的关键,其反应的难易程度取决于氢转移的能垒。此外,图 5还分别展示了H、OH和CH3这3种自由基夺取丙烯链端甲基时速率常数随温度的变化情况。与夺取丙烷链端甲基时的情况类似,H自由基的效果最佳,3个反应从易到难分别为路径r2-16、r2-17、r2-18,但与夺取甲基上的氢原子[4, 13, 43]相比,依旧有至少6个数量级的差距。
图 6和7展示了丙炔在不同反应条件下的能垒和速率常数的分布情况。由3.1节的介绍可知,丙炔与水蒸气加成可以生成CH3COHCH2或CH3CHCHOH,随后通过2次氢转移实现中间产物的裂解。根据图 6展示的路径r3-1到r3-4的能垒分布情况可以发现丙炔与水蒸气反应的关键步骤都是第一步(加成反应),后续的氢转移过程反而所需能量较小。因此,丙炔和H2O的加成反应过程决定了整个裂解反应的速率常数,无论CH3COHCH2后续如何通过氢转移实现进一步裂解(路径r3-1到r3-3),相应的反应速率常数都相同。如图 7所示,路径r3-4是4个反应路径中最容易发生的,相应的速率常数从600 ℃的9.309×10−6增至1 000 ℃的1.831×10−1(L·mol−1·s−1)。同时,结合前面的分析可以看出,丙炔是3种C3链烃中最容易在水蒸气作用下裂解的。路径r3-4的速率常数比相同温度下路径r1-4(丙烷)和r2-5(丙烯)分别高出9~13和3~4个数量级。

|
图 6 丙炔的能垒 Fig.6 Energy barriers of propyne |

|
图 7 丙炔在不同反应路径下的速率常数(L·mol−1·s−1) Fig.7 Reaction rate constants of propyne under different reaction paths(L·mol−1·s−1) |
当O自由基参与丙炔的裂解反应时,反应速率常数最大的是路径r3-7和r3-8。虽然初始的加成产物不同,但是这2个路径的关键步骤都对应中间产物CH3CHCO进一步裂解生成CH2CH2和CO,因此二者具有相同的速率常数。这也进一步说明在O自由基和丙炔反应时,无论一开始O自由基是与哪一个三键碳原子结合,最终都不会影响反应的速率常数。同时,如路径r3-5和r3-6所示,O自由基也能有效实现丙炔裂解,相应的速率常数与路径r3-7和r3-8的速率常数差值不超过1个数量级。
类似的,H自由基参与丙炔的裂解反应时首先进行加成反应,从图 6中的能垒分布情况来看,H自由基与中间三键碳原子结合所需的能量略高,但是在后续的裂解反应中,加成产物CH3CHCH的进一步裂解是相对容易实现的。如图 7所示,路径r3-11的速率常数是4个反应中最大的,随着温度从600升高到1 000 ℃,相应的速率常数从4.197×107增加到4.849×1010(L·mol−1·s−1),这与Peukert等[44]的总体趋势相近,但由于反应压力的不同以及实验过程中各种反应之间的交互影响,本研究算得的速率常数在600 ℃时比Peukert等[44]的结果低约2个数量级,而在1 000 ℃时则是高出5倍左右。同时,路径r3-11也是所有关于丙炔裂解反应路径中最容易发生的,结合前面的分析可以看出,H自由基在丙烯或者丙炔的裂解过程中起着决定性的作用。此外,在H自由基参与丙炔裂解的反应路径中,路径r3-10也是十分重要的。从整个反应流程来看,路径r3-10需要先将CH3CCH2转化成CH3CHCH才能进行后续的裂解反应,但是相应的氢转移过程所需能量较高,是整个反应的决速步,从而导致路径r3-10的速率常数要比相同温度下路径r3-11速率常数低3~4个数量级。从图 7中也可以看出,路径r3-9和r3-12具有相同的反应速率常数并且都是较难进行的反应路径,这主要是因为中间产物CH3CCH2很难直接脱除链端的甲基。
从OH自由基与丙炔反应时能垒的分布情况来看(图 6),3种反应路径的决速步都是第三步反应。在此基础上算得相应的速率常数由大到小分别是路径r3-15、r3-14和r3-13。其中,路径r3-15的速率常数从600 ℃的1.355×105增加到1 000 ℃的3.990×108(L·mol−1·s−1)。路径r3-14的速率常数则比相同温度下路径r3-15的速率常数低1个数量级。此外,由CH2CHCO进一步脱除CO也是可以实现的,但是在此之前,醛基中的氢和链端甲基中的氢彼此结合促使CH3CHCHO转化为CH2CHCO的过程较难实现,这也导致路径r3-13的速率常数比相同温度下路径r3-15的速率常数低5~7个数量级。但是从图 7中也可以看出,随着温度升高,路径r3-13的速率常数有了更为明显的提升,逐渐缩小了同另外2个反应路径的差距。
图 7中还展示了3种自由基在夺取链端甲基时速率常数随温度的变化情况。其中,丙炔与H自由基的反应是最容易进行的。但是随着链烃的不饱和度增加,H和OH自由基直接夺取甲基的难度也在逐步上升。与夺取甲基上的氢原子[3, 45]相比,相应的速率常数低了至少7个数量级。
4 结论采用Gaussian及其配套软件,从微观的角度对C3系列链烃化合物在H2O或O/H/OH/CH3自由基作用下的裂解反应过程进行了分析。从整体来看,丙烷是一种典型的饱和短链烃,大多数可以通过单步反应实现裂解。其中,H/OH/CH3自由基可以直接攻击丙烷端链上的甲基来实现链烃脱碳。但是丙烯和丙炔的裂解过程往往涉及多个异构体之间的转换,具有较为复杂的反应网络。H2O提供的O/H/OH自由基在与2种不饱和烃(丙烯及丙炔)反应时,通常是先在双键或者三键位置进行加成,然后通过旋转和结构重排促使中间产物进一步裂解。
在本研究中,H/OH自由基相对于O自由基而言,更有助于丙烷、丙烯及丙炔的裂解,其中能够实现脱碳过程的最优反应均是链烃与H自由基的反应,并且速率常数与不饱和度成正比,因此H2O作为H/OH自由基的直接供体,有利于C3系列链烃化合物的裂解。
| [1] |
WONG S L, NGADI N, ABDULLAH T A T, et al. Current state and future prospects of plastic waste as source of fuel: A review[J]. Renewable and Sustainable Energy Reviews, 2015, 50: 1167-1180. DOI:10.1016/j.rser.2015.04.063 |
| [2] |
SANSANIWAL S K, PAL K, ROSEN M A, et al. Recent advances in the development of biomass gasification technology: A comprehensive review[J]. Renewable and Sustainable Energy Reviews, 2017, 72: 363-384. DOI:10.1016/j.rser.2017.01.038 |
| [3] |
ROSADO-REYES C M, MANION J A, TSANG W. Kinetics of the thermal reaction of h atoms with propyne[J]. The Journal of Physical Chemistry A, 2010, 114(18): 5710-5717. DOI:10.1021/jp9122858 |
| [4] |
ROSADO-REYES C M, MANION J A, TSANG W. H atom attack on propene[J]. The Journal of Physical Chemistry A, 2011, 115(13): 2727-2734. DOI:10.1021/jp111476q |
| [5] |
VANUZZO G, BALUCANI N, LEONORI F, et al. Reaction dynamics of O(3P) + propyne: I. Primary products, branching ratios, and role of intersystem crossing from crossed molecular beam experiments[J]. The Journal of Physical Chemistry A, 2016, 120(27): 4603-4618. DOI:10.1021/acs.jpca.6b01563 |
| [6] |
BALUCANI N, LEONORI F, NEVRLY V, et al. Reaction dynamics and relative yields of the H- and CH3-displacement channels in the O+CH3CCH reaction[J]. Chemical Physics Letters, 2014, 602: 58-62. DOI:10.1016/j.cplett.2014.04.016 |
| [7] |
VASU S S, HONG Z, DAVIDSON D F, et al. Shock tube/laser absorption measurements of the reaction rates of OH with ethylene and propene[J]. The journal of physical chemistry. A, 2010, 114(43): 11529-11537. DOI:10.1021/jp106049s |
| [8] |
BADRA J, NASIR E F, FAROOQ A. Site-specific rate constant measurements for primary and secondary H- and D-abstraction by OH radicals: Propane and n-butane[J]. The Journal of Physical Chemistry A, 2014, 118(26): 4652-4660. DOI:10.1021/jp503849b |
| [9] |
张红梅, 林枫, 任铭琪, 等. 小分子烃类蒸汽热裂解自由基机理模型研究方法的探讨[J]. 化工学报, 2017, 68(4): 1423-1433. ZHANG H M, LIN F, REN M Q, et al. Free radical models of small molecular alkane pyrolysis[J]. CIESC Journal, 2017, 68(4): 1423-1433. |
| [10] |
WANG B, HOU H, GU Y. Mechanism and rate constant of the reaction of atomic hydrogen with propyne[J]. The Journal of Chemical Physics, 2000, 112(19): 8458-8465. DOI:10.1063/1.481484 |
| [11] |
DARANLOT J, BERGEAT A, CARALP F, et al. Gas-phase kinetics of hydroxyl radical reactions with alkenes: Experiment and theory[J]. ChemPhysChem, 2010, 11(18): 4002-4010. DOI:10.1002/cphc.201000467 |
| [12] |
IZSÁK R, SZŐRI M, KNOWLES P J, et al. High accuracy ab initio calculations on reactions of OH with 1-alkenes. The case of propene[J]. Journal of Chemical Theory and Computation, 2009, 5(9): 2313-2321. DOI:10.1021/ct900133v |
| [13] |
HAN P, SU K, LIU Y, et al. Reaction rate of propene pyrolysis[J]. Journal of Computational Chemistry, 2011, 32(13): 2745-2755. DOI:10.1002/jcc.21854 |
| [14] |
GIMONDI I, CAVALLOTTI C, VANUZZO G, et al. Reaction dynamics of O(3P) + propyne: II. Primary products, branching ratios, and role of intersystem crossing from ab initio coupled triplet/singlet potential energy surfaces and statistical calculations[J]. The Journal of Physical Chemistry A, 2016, 120(27): 4619-4633. DOI:10.1021/acs.jpca.6b01564 |
| [15] |
BUETTNER A D, DILDAY B J, CRAIGMILE R A, et al. The reaction of O(3P) with alkynes: A dynamic and computational study focusing on formyl radical production[J]. Physical Chemistry Chemical Physics, 2020, 22(42): 24583-24599. DOI:10.1039/D0CP03698F |
| [16] |
LEONORI F, BALUCANI N, NEVRLY V, et al. Experimental and theoretical studies on the dynamics of the O(3P) + propene reaction: primary products, branching ratios, and role of intersystem crossing[J]. The Journal of Physical Chemistry C, 2015, 119(26): 14632-14652. DOI:10.1021/jp512670y |
| [17] |
PARANDAMAN A, RAJAKUMAR B. Addition and abstraction kinetics of H atom with propylene and isobutylene between 200 and 2500K: A DFT study[J]. Chemical Physics, 2017, 491: 82-94. DOI:10.1016/j.chemphys.2017.05.008 |
| [18] |
OGLIARO F, BEARPARK M J, HEYD J J, et al. Gaussian 16, Revision C. 01 [CP]. Wallingford: Gaussian Inc, 2016.
|
| [19] |
LU T, CHEN Q. Shermo: A general code for calculating molecular thermochemistry properties[J]. Computational and Theoretical Chemistry, 2021, 113249. |
| [20] |
MUSIAŁ M, BARTLETT R J. Coupled-cluster theory in quantum chemistry[J]. Reviews of Modern Physics, 2007, 79(1): 291-352. DOI:10.1103/RevModPhys.79.291 |
| [21] |
杨振丽. 烷基过氧自由基和芳香烃双环过氧自由基与HO2的化学反应动力学理论研究[D]. 合肥: 中国科学技术大学, 2020. YANG Z L. A theoretical study on the chemical reaction kinetics of the alkyl peroxy radicals and aromatic bicyclic peroxy radicals with HO2 reactions [D]. Hefei: University of Science and Technology of China, 2020. |
| [22] |
BOMBLE Y J, STANTON J F, KÁLLAY M, et al. Coupled-cluster methods including noniterative corrections for quadruple excitations[J]. The Journal of Chemical Physics, 2005, 123(5): 54101. DOI:10.1063/1.1950567 |
| [23] |
KÁLLAY M, GAUSS J. Approximate treatment of higher excitations in coupled-cluster theory[J]. The Journal of Chemical Physics, 2005, 123(21): 214105. DOI:10.1063/1.2121589 |
| [24] |
DAVIDSON E R. Comment on "Comment on Dunning's correlation-consistent basis sets"[J]. Chemical Physics Letters, 1996, 260(3): 514-518. |
| [25] |
MERRICK J P, MORAN D, RADOM L. An evaluation of harmonic vibrational frequency scale factors[J]. The Journal of Physical Chemistry A, 2007, 111(45): 11683-11700. DOI:10.1021/jp073974n |
| [26] |
SKODJE R, TRUHLAR D, GARRETT B. Vibrationally adiabatic models for reactive tunneling[J]. Journal of Chemical Physics, 1982, 77(12): 5955-5976. DOI:10.1063/1.443866 |
| [27] |
GONZALEZ C, SCHLEGEL H B. Reaction path following in mass-weighted internal coordinates[J]. The Journal of Physical Chemistry, 1990, 94(14): 5523-5527. DOI:10.1021/j100377a021 |
| [28] |
SIVARAMAKRISHNAN R, SU M C, MICHAEL J V, et al. Shock tube and theoretical studies on the thermal decomposition of propane: evidence for a roaming radical channel[J]. The Journal of Physical Chemistry A, 2011, 115(15): 3366-3379. DOI:10.1021/jp2006205 |
| [29] |
OEHLSCHLAEGER M A, DAVIDSON D F, HANSON R K. High-temperature ethane and propane decomposition[J]. Proceedings of the Combustion Institute, 2005, 30(1): 1119-1127. DOI:10.1016/j.proci.2004.07.032 |
| [30] |
ZHU R S, XU Z F, LIN M C. Ab initio studies of alkyl radical reactions: Combination and disproportionation reactions of CH3 with C2H5, and the decomposition of chemically activated C3H8[J]. The Journal of Chemical Physics, 2004, 120(14): 6566-6573. DOI:10.1063/1.1665370 |
| [31] |
MOUSAVIPOUR S H, HOMAYOON Z. A theoretical study on the kinetics of disproportionation versus association reaction of CH3 + C2H5[J]. The Journal of Physical Chemistry A, 2003, 107(41): 8566-8574. DOI:10.1021/jp030597f |
| [32] |
HUNG W, TSAI C, MATSUI H, et al. Experimental and theoretical study on the thermal decomposition of C3H6 (propene)[J]. The Journal of Physical Chemistry A, 2015, 119(8): 1229-1237. DOI:10.1021/jp5102169 |
| [33] |
NGUYEN T L, PEETERS J, VEREECKEN L. Quantum chemical and statistical rate study of the reaction of O(3P) with allene: O-addition and H-abstraction channels[J]. The Journal of Physical Chemistry A, 2006, 110(44): 12166-12176. DOI:10.1021/jp0639905 |
| [34] |
HU W, LENDVAY G, MAITI B, et al. Trajectory surface hopping study of the O(3P) + ethylene reaction dynamics[J]. The Journal of Physical Chemistry A, 2008, 112(10): 2093-2103. DOI:10.1021/jp076716z |
| [35] |
NGUYEN T L, VEREECKEN L, HOU X J, et al. Potential energy surfaces, product distributions and thermal rate coefficients of the reaction of O(3P) with C2H4 (XAg): A comprehensive theoretical study[J]. The Journal of Physical Chemistry A, 2005, 109(33): 7489-7499. DOI:10.1021/jp052970k |
| [36] |
TALOTTA F, MORISSET S, ROUGEAU N, et al. Electronic structure and excited states of the collision reaction O(3P) + C2H4: A multiconfigurational perspective[J]. The Journal of Physical Chemistry A, 2021, 125(28): 6075-6088. DOI:10.1021/acs.jpca.1c02923 |
| [37] |
BLITZ M A, BEASLEY M S, PILLING M J, et al. Formation of the propargyl radical in the reaction of 1CH2 and C2H2: Experiment and modelling[J]. Physical Chemistry Chemical Physics, 2000, 2: 805-812. DOI:10.1039/a907959i |
| [38] |
MILLER J A, KLIPPENSTEIN S J. From the multiple-well master equation to phenomenological rate coefficients: Reactions on a C3H4 potential energy surface[J]. The Journal of Physical Chemistry A, 2003, 107(15): 2680-2692. DOI:10.1021/jp0221082 |
| [39] |
SIVARAMAKRISHNAN R, MICHAEL J V, RUSCIC B. High-temperature rate constants for H/D + C2H6 and C3H8[J]. International Journal of Chemical Kinetics, 2012, 44(3): 194-205. DOI:10.1002/kin.20607 |
| [40] |
COHEN N. Are reaction rate coefficients additive? Revised transition state theory calculations for OH + alkane reactions[J]. International Journal of Chemical Kinetics, 1991, 23(5): 397-417. DOI:10.1002/kin.550230506 |
| [41] |
BUXTON G V, GREENSTOCK C L, HELMAN W P, et al. Critical review of rate constants for reactions of hydrated electrons, chemical kinetic data base for combustion chemistry. part 3: Propane[J]. Journal of Physical and Chemical Reference Data, 1988, 17(2): 536-635. |
| [42] |
HIDAKA Y, NAKAMURA T, TANAKA H, et al. Shock tube and modeling study of propene pyrolysis[J]. International Journal of Chemical Kinetics, 1992, 24(9): 761-780. DOI:10.1002/kin.550240902 |
| [43] |
TSANG W. Chemical kinetic data base for combustion chemistry part V: Propene[J]. Journal of Physical & Chemical Reference Data, 1991, 20(2): 221-273. |
| [44] |
PEUKERT S L, LABBE N J, SIVARAMAKRISHNAN R, et al. Direct measurements of rate constants for the reactions of CH3 radicals with C2H6, C2H4, and C2H2 at high temperatures[J]. The Journal of Physical Chemistry A, 2013, 117(40): 10228-10238. DOI:10.1021/jp4073153 |
| [45] |
HANSEN N, MILLER J A, WESTMORELAND P R, et al. Isomer-specific combustion chemistry in allene and propyne flames[J]. Combustion and Flame, 2009, 156(11): 2153-2164. DOI:10.1016/j.combustflame.2009.07.014 |