




2. 百色学院 化学与环境工程学院,广西 百色 533000
2. College of Chemistry and Environment Engineering, Baise University, Baise 533000, China
四氢噻吩(Tetrahydrothiophene,THT),是一种重要的含硫饱和杂环化合物,主要作为煤气和天然气的加臭剂[1, 2]。四氢噻吩还可以作为链转移抑制剂、改性剂以及合成多种新型医药、农药及高分子材料助剂的中间体等[3]。相对于传统的四氢噻吩合成工艺—噻吩催化加氢法、四氢呋喃直接硫代法及1, 4二氯丁烷硫代法[4~6],以含硫氧化合物环丁砜选择性加氢脱氧制取四氢噻吩的技术路线,则具有原料价廉易得、过程安全且无环境污染、目的产物产率高等优点[7]。该反应过程的化学反应方程式如下:
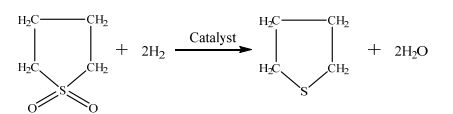
|
与Ni-Mo负载型催化剂相比,本体型Ni-Mo催化剂因其可以提供相对较多的加氢反应活性中心,因而在较缓和的条件下也可具有优良的加氢性能。为避免高温下环丁砜的分解,以使环丁砜选择性加氢脱氧在较缓和的条件下进行,宜采用高活性的本体型Ni-Mo催化剂。在前期的研究工作中发现,环丁砜选择性加氢脱氧中本体型Ni-Mo催化剂存在缓慢失活和目的产物选择性下降的问题[8]。本文在前期研究工作的基础上,采用多种技术手段表征失活催化剂,研究了催化剂的失活机理,进而实验确定了适宜的失活催化剂再生条件,以期为环丁砜选择性加氢脱氧生产四氢噻吩过程的工业化提供技术支持。
2 实验部分 2.1 实验材料钼酸铵((NH4)6Mo7O24·4H2O,分析纯)、硝酸镍(Ni(NO3)2·6H2O,分析纯)、柠檬酸(C6H8O7·H2O,分析纯)、无水乙醇(分析纯)、氨水(分析纯)、N, N-二甲基甲酰胺(分析纯)和乙基苯(分析纯)均由天津市科密欧化学试剂有限公司提供;氢氧化铝干胶由山东公泉化工股份有限公司提供;田菁粉由黑龙江大庆化工研究中心提供;环丁砜(C4H8O2S,分析纯)由天津市光复化工研究所提供;四氢噻吩(C4H8S,分析纯)由河北辛集市顺隆化工有限公司提供;氢气(纯度99.99%)由天津市淦达气体有限公司提供。
2.2 催化剂的制备采用溶胶凝胶法制备本体型Ni-Mo催化剂。将钼酸铵、硝酸镍、柠檬酸(三者摩尔比为0.57:5:9) 以及一定量的N, N-二甲基甲酰胺,溶解于含乙醇体积分数50%的水溶液中,搅拌均匀后用氨水调节溶液的pH,在80℃水浴中加热直至溶液形成湿凝胶,将湿凝胶经超声振荡、老化、干燥处理后得到干凝胶。将所得干凝胶于管式炉中氮气保护下260℃预焙烧,除去柠檬酸后再在马弗炉中于500℃焙烧3 h,即得Ni-Mo复合氧化物粉体。将Ni-Mo复合氧化物粉体、黏结剂氢氧化铝干胶和助挤剂田菁粉按质量比7:3:0.3混合研磨均匀,以质量分数3%的稀硝酸为胶溶剂,采用机械混捏法挤条成型,焙烧后得本体型Ni-Mo催化剂。
2.3 催化剂的表征催化剂的晶相结构分析在德国Bruker公司的达芬奇型X射线粉末衍射仪上进行,条件为Cu靶K辐射、工作电流40 mA、电压40 kV、扫描范围5~70°。催化剂表面元素组成在美国EDAX公司的PV9900型X-射线能谱仪上进行分析。催化剂的孔结构性质分析在美国Micromeritics公司生产的ASPA2020型比表面积和孔隙度分析仪上进行,样品在423 K下抽真空,脱附4 h,在液氮温度下吸附N2,根据催化剂的N2吸附脱附特性,获得催化剂的比表面积(BET法)和孔径分布(BJH法,脱附分支)数据。催化剂积炭程度的同步热分析在美国TA公司生产的SDT-2960型DSC-TGA同步一体机上进行,分析条件为空气氛围,以10℃·min-1的升温速率由室温升至900℃。采用美国Perkin Elmer公司的Opfima 7300 Ⅴ型的电感耦合等离子发射光谱仪测定催化剂的Mo、Ni等元素含量。采用荷兰Philips公司生产的ESEM EMXL-30扫描电子显微镜观测催化剂的表面形貌,观测前对样品表面进行喷金处理,仪器的工作电压为15 kV。
2.4 催化剂的评价乙基苯对环丁砜具有很好的互溶性,且与产物四氢噻吩有一定的沸点差而易于分离。前期的实验研究表明,在反应过程中乙基苯基本为惰性的。因此在本研究工作中仍采用含质量分数10%环丁砜的乙基苯溶液为原料评价催化剂的性能。催化剂的评价在固定床反应装置上进行,反应器长约340 mm,外径19 mm,内径10 mm;催化剂颗粒长约2~3 mm,直径2 mm,装填量5 mL,催化剂两端填充粒径20~40目石英砂。反应前催化剂在常压、H2氛围下,以10℃·min-1的升温速率升温至400℃,恒温3 h还原后,当温度降至反应温度230℃时,调节反应压力为0.3 MPa,液时空速为1 h-1,氢气和原料的体积比为200,用高压微进样泵将原料泵入反应器进行反应。冷凝后的反应产物组成分析在北分瑞利公司生产的SP-3420A气相色谱仪上进行,毛细管柱AC1(50 m×0.32 mm×0.5 μm),氢火焰检测器。基于本文的研究目的,以环丁砜的转化率和四氢噻吩的选择性来评价催化剂的催化性能。
3 结果与讨论 3.1 催化剂的稳定性反应压力0.3 MPa,温度230℃,V氢气/V原料= 200,液时空速1 h-1条件下,在本体型Ni-Mo催化剂上,环丁砜选择性加氢脱氧反应的环丁砜转化率和四氢噻吩选择性随时间的变化见图 1。

|
图 1 环丁砜转化率和四氢噻吩选择性随时间的变化趋势 Fig.1 Sulfolane conversion and THT selectivity as a function of time |
由图 1可知,从反应初始至前200 h内,环丁砜的转化率和四氢噻吩的选择性分别保持在96%与94%左右,即催化剂的催化性能较为稳定;在反应200 h后可以看到,虽然环丁砜的转化率基本维持在96%不变,而四氢噻吩的选择性呈现明显的下降趋势,即催化剂的失活问题已显现。
3.2 本体型Ni-Mo催化剂的失活原因分析 3.2.1 催化剂的XRD表征及EDS分析为研究反应过程中催化剂晶型结构和表面元素组成变化对其催化性能的影响,将新鲜催化剂(还原态)及反应72和600 h后的催化剂分别记作Cat-000、Cat-072、Cat-600,三个催化剂的XRD表征结果见图 2;Cat-072、Cat-600的表面主要元素组成的EDS分析结果见表 1。

|
图 2 新鲜及反应后催化剂的XRD谱图 Fig.2 XRD patterns of fresh and used catalysts |
| 表 1 反应后催化剂表面主要元素的质量分数 Table 1 Mass fractions of major elements on the surface of used catalysts |
由图 2可以看出,新鲜(还原态)催化剂Cat-000的主要衍射峰为MoO2、MoNi与少量的NiMoO4;而反应72 h后的Cat-072则在2θ分别为14.3、32.8和21.8°、31.1、37.8、44.4、50.2和55.8°等处出现了MoS2、Ni3S2的衍射峰[9];随着反应的延长,Cat-600的MoS2的衍射峰已基本消失。据此可知,随着反应时间的增加,催化剂的晶型结构发生了变化。从环丁砜选择性加氢脱氧的反应机理可知,在环丁砜加氢脱氧生成四氢噻吩的过程中,不可避免地有少量四氢噻吩被进一步加氢脱硫而生成H2S[10]。这些生成的H2S极易选择性地化学吸附在催化剂表面,与催化剂中的活性组分MoO2和Ni结合形成具有较高加氢脱硫活性的MoS2和Ni3S2相[11, 12]。从表 1催化剂表面主要元素的EDS能谱分析中可以看出,与催化剂Cat-072相比,催化剂Cat-600表面的S和Ni元素含量显著增加,可以推测出随着反应时间的延长催化剂的硫化程度加深,且发生了催化剂内部的Ni原子向催化剂表面迁移的现象,迁移的Ni原子进一步进入MoS2的层状堆积结构而形成了Ni-Mo-S相,进而使得MoS2的衍射峰强度逐渐降低以至消失[13]。研究表明Ni-Mo-S活性相是加氢脱硫的活性中心,此类活性中心的生成将促进四氢噻吩的加氢脱硫反应[14, 15],因此使得反应过程中四氢噻吩的选择性逐渐下降。
3.2.2 催化剂的DSC-TGA分析热分析是研究催化剂积炭失活的主要手段。为了考察反应过程中积炭对催化剂催化性能的影响,分别对反应72 h及600 h的催化剂做了DSC-TGA分析,分析结果分别见图 3(a)和图 3(b)。

|
图 3 反应72 h及600 h催化剂的DSC-TGA曲线 Fig.3 DSC-TGA curves of the catalysts used for 72 h and 600 h |
从图 3可以看出,反应后的催化剂在空气氛围下大致存在4个失重区,分别出现在25~250、300~425、430~550和650~900。25~250℃的失重可以归结为催化剂上残留原料油的挥发和物理吸附水的脱附;300~425℃的失重可归结为Ni3S2、MoS2的氧化燃烧引起的[16, 17],Girard等[18]在用DSC-TGA分析技术研究单金属氧化物催化剂中毒再生时发现,MoS2在400℃附近有明显的失重及显著的放热峰,且在此失重范围内SO2的释放量明显增加。因此对应本研究的DSC-TGA曲线上可以看出在此失重区间内Cat-072的硫含量约为0.46%,而Cat-600的硫含量约为1.96%。根据图 3中DSC-TGA曲线在500℃附近存在一个明显的失重和放热峰及催化剂在反应过程中其表面易存在不易被氧化的假石墨型积炭的情形,因此430~550℃的失重可归结为催化剂上积炭的脱除。对于此部分的积炭,从3(a)和3(b)上可以看到,Cat-072约为1.41%,而Cat-600约为3.84%。此外由于MoS2在空气中燃烧生成MoO3且MoO3具有易升华的特点[18],因此可推测650~900℃的失重应为MoO3的升华。结合3.1节中催化剂600 h的稳定性评价结果及上述DSC-TGA曲线的积炭分析可知,随着反应时间的延长,虽然催化剂上的积炭量有所增加,但环丁砜的转化率并没有出现下降的趋势。据此可推测,积炭不是导致催化剂催化性能下降的主要原因。
3.2.3 催化剂的孔结构分析新鲜(还原态)催化剂及反应600 h后催化剂的吸附-脱附等温曲线和孔径分布曲线分别见图 4(a)和图 4(b),相应的催化剂孔结构参数见表 2。

|
图 4 新鲜及反应600 h后催化剂的N2吸附-脱附等温曲线和孔径分布曲线 Fig.4 N2 adsorption and desorption profiles and the pore size distribution of fresh and used catalysts |
| 表 2 新鲜及反应600 h催化剂的孔结构参数 Table 2 Texture parameters of the fresh and used catalysts |
从图 4(a)、(b)中可以看出,催化剂失活前后的N2吸附-脱附等温曲线前后均呈现IUPAU分类中的Ⅳ型吸附等温线,在相对压力较高处存在一滞后环,这表明失活前后的催化剂均具有介孔结构,且催化剂的最可几孔径变化较小,均为3.6 nm左右。从表 2给出的催化剂孔结构参数可知,与新鲜(还原态)催化剂相比,反应600 h后催化剂的比表面积、孔体积和平均孔径分别下降了18.5%、36.1%和5.0%。积炭应是导致上述催化剂孔结构参数明显下降的主要原因。催化剂上积炭的来源可能由于随着反应的进行吸附在催化剂表面的产物四氢噻吩发生分解,生成易聚合的含碳原子团,停留在催化剂表面将金属包埋并形成积炭[19],桑小义等[20]在研究含氧化合物加氢脱氧时发现,烯烃、芳香烃等极易吸附在催化剂表面,进而聚合形成积炭,堵塞催化剂孔道,造成催化剂的比表面积、孔体积和平均孔径下降。由于本体型Ni-Mo催化剂加氢脱氧的活性中心密度较大,且其被逐渐硫化后形成的Ni-Mo-S相的加氢脱氧、脱硫活性更高,所以在积炭程度较低时催化剂的加氢活性并未受到影响。
3.2.4 催化剂的ICP-AES分析新鲜(还原态)、反应72 h及600 h的催化剂的ICP-AES元素分析结果见表 3。从表 3可以看出,失活前后催化剂的Mo/Ni摩尔比均在0.84左右,与所制备催化剂的投料Mo/Ni摩尔比0.80基本一致,这表明失活催化剂并未出现活性组分的流失。与Cat-000相比,Cat-600的Mo、Ni含量分别减少了20.0%左右,这应是由于催化剂表面生成较多的积炭和部分还原态的活性组分转化为硫化态等所致。
| 表 3 新鲜、反应72 h及600 h催化剂的金属元素分布 Table 3 The distribution of metal elements in the fresh and used catalysts |
图 5为新鲜(还原态)及反应600 h的本体型Ni-Mo催化剂的SEM表征结果。

|
图 5 新鲜及反应600 h后催化剂样品的SEM谱图 Fig.5 SEM micrographs of fresh and used catalysts |
由图 5可知,新鲜催化剂的孔道较为丰富,其表面的活性组分颗粒分布相对均匀,而反应600 h催化剂的孔道数量则明显减少,这可能是由于积炭覆盖所致。比较图 5(a)和(b)未见Cat-600的颗粒尺寸与Cat-000的颗粒尺寸有显著的差异,这表明随反应时间的增加催化剂未出现明显的烧结及颗粒团聚现象。
3.3 催化剂的再生从3.1节中可知,在环丁砜选择性加氢脱氧过程中本体型Ni-Mo催化剂的失活较为缓慢,为便于研究失活催化剂的再生,首先采用较为苛刻的反应条件并以含硫产物四氢噻吩为原料对催化剂进行加速硫化和积炭以得到失活催化剂。加速硫化和积炭的原料为含质量分数10%四氢噻吩的乙基苯溶液,反应条件为反应温度300℃、压力1 MPa、空速1 h-1和氢油比200,加速失活时间15 h。失活催化剂的催化性能随反应时间的变化见图 8。以空气为再生介质,对失活催化剂进行原位烧硫烧炭再生,空气流量为50 mL·min-1,再生程序为以2℃·min-1升温速率由室温升温到400℃,恒温2 h进行失活催化剂的脱油和烧硫。再以2℃·min-1的升温速率,升温到500℃恒温3 h,以脱除催化剂的表面积炭。将再生后催化剂记为Cat-R,其DSC-TGA曲线见图 6。新鲜(氧化态)催化剂记为Cat-O,其与Cat-R的XRD谱图见图 7。再生后催化剂Cat-R的催化性能随反应时间的变化趋势见图 9。

|
图 8 失活催化剂的催化性能随反应时间的变化趋势 Fig.8 Catalytic performance of the deactivated catalysts as a function of time |

|
图 6 再生催化剂的DSC-TGA曲线 Fig.6 DSC-TGA curves of the regenerated catalyst |

|
图 7 新鲜及再生后催化剂的XRD谱图 Fig.7 XRD patterns of the fresh and regenerated catalysts |

|
图 9 再生催化剂的催化性能随反应时间的变化趋势 Fig.9 Catalytic performance of the regenerated catalyst as a function of time |
由图 6可以看出,加速失活催化剂经空气程序升温烧硫烧炭再生后,其DSC-TGA曲线中并没有出现显著的硫、碳的放热峰,表明表面的积炭积硫基本脱除。而图 7中新鲜(氧化态)及加速失活再生后催化剂的XRD谱图可以看出,再生后催化剂的表观物相与新鲜(氧化态)催化剂一致,活性相得以有效恢复均为NiMoO4晶相,说明失活催化剂易于氧化再生,且再生后催化剂表面没有出现金属活性组分聚集的现象。
由图 8可知,经加速失活后催化剂的催化性能显著下降,环丁砜的平均转化率降低为49%,四氢噻吩的平均选择性降低为26%。从图 9可看到,经空气程序升温烧硫烧炭后,催化剂的催化性能得以显著恢复,环丁砜的转化率约为93%,四氢噻吩的选择性约为87%,二者分别恢复到新鲜催化剂的97%和93%。这表明用于环丁砜选择性加氢脱氧过程的本体型Ni-Mo催化剂具有优异的再生性能。
4 结论在催化环丁砜选择性加氢脱氧过程中,本体型Ni-Mo催化剂的催化性能存在缓慢的失活(主要是目的产物THT选择性下降)现象。DSC-TGA、N2-BET、ICP-AES、SEM等表征结果表明,失活催化剂没有出现活性组分的流失及烧结问题;积炭虽造成催化剂的比表面积、孔径和孔体积明显下降,但其对催化剂性能的影响较小;催化剂中的Mo、Ni活性组分在反应过程中与副产物H2S结合生成了MoS2、Ni3S2相,随着反应时间的延长,催化剂上的硫含量逐渐增加,进而导致具有较强加氢脱硫活性的Ni-Mo-S活性相形成,因而导致催化剂的选择性下降。烧硫烧炭再生可有效恢复失活催化剂的催化性能。
| [1] | ZHAO Da-sheng(赵大生), LIU Hai-yan(刘海燕). New synthsis process of tetrahydrothiophene(四氢噻吩合成新工艺)[J]. Heilongjiang Science(黑龙江科学) , 2010(2): 23-25. |
| [2] | ZHU Zhi-qiang(朱志强). The application and importance of natural gas and odor agent in the life of residents(天然气加臭剂在居民生活用气中的用途及重要性)[J]. Science Technology Innovation and Application(科技创新与应用) , 2013(36): 49-49. |
| [3] | LI Pi-gao(李丕高), LI Gang(李刚). Improvement on the synthesis of tetrahydrothiophene(四氢噻吩合成方法的改进)[J]. Chinese Journal of Synthetic Chemistry(合成化学) , 2007, 15(3): 374-375. |
| [4] | LI Hong-bin(李鸿滨). Production technology and market analysis of tetrahydrothiophene(四氢噻吩的生产技术与市场分析)[J]. Chemical Intermediate(化工中间体) , 2006(6): 12-19. |
| [5] | Ermakova A, Mashkina A V, Sakhaltueva L G. Kinetic study of catalytic hydrogenation of thiophene on a palladium sulfide catalyst[J]. Kinetics and Catalysis , 2002, 43(4): 528-536. DOI:10.1023/A:1019831203005. |
| [6] | Keith L J, Easley W K, Wagner W S. Tetrahydrothiophene[M].New York: John Wiley & Sons, 2003. |
| [7] | DUAN Yan(段艳), WANG Xin(王欣), HOU Kai-hu(侯凯湖). Preparation and evaluation of unsupported Ni-Mo catalysts for synthesis of tetrahydrothiophene(合成四氢噻吩非负载Ni-Mo催化剂的制备与表征)[J]. Chemical Reaction Engineering and Technology(化学反应工程与工艺) , 2016, 32(2): 164-169. |
| [8] | DUAN Yan(段艳), WANG Xin(王欣), HOU Kai-hu(侯凯湖). Effects of reduction conditions on unsupported Ni-Mo composite oxide catalysts for selective hydrodeoxygenation(还原条件对Ni-Mo非负载催化剂选择性加氢脱氧的影响)[J]. Journal of Chemical Engineering of Chinese Universities(高校化学工程学报) , 2016, 30(6): 1451-1457. |
| [9] | Yin C, Zhao L, Bai Z, et al. A novel porous ammonium nickel molybdate as the catalyst precursor towards deep hydrodesulfurization of gas oil[J]. Fuel , 2013, 107(9): 873-878. |
| [10] | Wang H, Enrique I. Mechanism and site requirements of thiophene hydrodesulfurization catalyzed by supported Pt clusters[J]. Chemcatchem , 2011, 3(7): 1166-1175. DOI:10.1002/cctc.v3.7. |
| [11] | Struis R P W J, Schildhauer T J, Czekaj I, et al. Sulphur poisoning of Ni catalysts in the SNG production from biomass:ATPO/XPS/XAS study[J]. Applied Catalysis A:General , 2009, 362(1-2): 121-128. DOI:10.1016/j.apcata.2009.04.030. |
| [12] | Marin-Flores O, Turba T, Ellefson C, et al. Sulfur poisoning of molybdenum dioxide during the partial oxidation of a Jet-A fuel surrogate[J]. Applied Catalysis , 2011, 105(1-2): 61-68. DOI:10.1016/j.apcatb.2011.03.035. |
| [13] | JIN Xin (金鑫). Preparation and catalytic properties of unsupported transition metal sulfides by thermal decomposition method (非负载过渡金属硫化物热解法制备及催化性能)[D]. Dalian (大连): Dalian University of Technology (大连理工大学), 2010. http://cdmd.cnki.com.cn/Article/CDMD-10141-2010111494.htm |
| [14] | Fujikawa T, Ribeiro F H, Somorjai G A. Hydrodesulfurization of tetrahydrothiophene over evaporated Mo, Co and Mo-Co model catalysts[J]. Catalysis Letters , 1999, 63(1): 21-26. |
| [15] | Mashkina A V. Thiophene hydrogenation to tetrahydrothiophene over tungsten sulfide catalysts[J]. Kinetics and Catalysis , 2003, 44(2): 277-282. DOI:10.1023/A:1023316831685. |
| [16] | LI Li-min(李利民), WANG Li-kui(王利魁). Promoters and sulfurizing degree of sulfurized WGS catalyst K-Ni-Mo/Al2O3(K-Ni-Mo/Al2O3耐硫变换催化剂硫化态与硫化度研究)[J]. Journal of Zhengzhou University (Natural Science Edition)((郑州大学学报(理学版))) , 2001, 33(4): 66-69. |
| [17] | SUN Wan-fu(孙万付), MA Bo(马波), SUO Ji-shuan(索继栓), et al. Study of coke deposition behavior of hydrocracking catalyst(加氢裂化催化剂积炭行研究)[J]. Chinese Journal of Catalysis(催化学报) , 2000, 21(3): 269-272. |
| [18] | Girard V, Baudot A, Chiche D, et al. Rational selection of single oxide sorbents for syngas desulfurization regenerable at reduced temperature:thermochemical alculations and experimental study[J]. Fuel , 2014, 128(21): 220-230. |
| [19] | JI Ke-ming(吉可明), MENG Fan-hui(孟凡会), LI Zhong(李忠). Recent advances in research on carbon deposition and deactivation of Ni supported catalysts(Ni负载催化剂积炭失活的研究进展)[J]. Natural Gas Chemical Industry (C1 Chemistry and Technology)(天然气化工(C1化学与化工)) , 2015(1): 83-88. |
| [20] | SANG Xiao-yi(桑小义), LI Hui-feng(李会峰), LI Ming-feng(李明丰), et al. Research advances of catalysts for the hydrodeoxygenation of oxygenic compounds(含氧化合物加氢脱氧催化剂研究进展)[J]. Acta Petrolei Sinica (Petroleum Processing Section)(石油学报(石油加工)) , 2014, 30(6): 1119-1127. |