




近年来,硝基吡唑类含能材料由于具有高生成热,良好热稳定性和爆轰性能,受到广泛关注[1-4]。多种硝基吡唑类含能化合物已经相继被合成。如,3, 4-二硝基吡唑[5-6],3, 5-二硝基吡唑(3, 5-DNP)[5, 7-8],3, 4, 5-三硝基吡唑[9-11],但对硝基吡唑氮氨化物的探究还较少。硝基吡唑的1位N上H比较活泼,因此在1位引入氨基等官能团成为可能[12]。正因为这个原因,可通过氨化试剂反应在1位N上引入氨基集团,形成分子内和分子间氢键,以提高化合物的热稳定性并降低感度;同时由于N-N键的存在,可以增加生成热。硝基吡唑氮氨化物还是重要的中间体,氨基可进一步形成硝氨基[12-13]、三硝基乙氨基[14]等含能基团。
目前针对硝基吡唑氨化常见氨化试剂有羟胺、O-磺酸羟胺、2, 4, 6-三甲基苯磺酰羟胺(MSH)、对甲基苯磺酰羟胺(TSH)等。其中羟胺、O-磺酸羟胺反应活性较低,常用于富电子氮杂环化合物胺化[6-7];MSH、TSH等反应活性较高,多用于含缺电子基团氮杂环化合物的氮胺化[8-9]。
ZHAO等[15]报道了1-氨基-3, 5-二硝基吡唑(ADNP)合成方法,以3, 5-DNP为原料,对甲基苯磺酰羟胺(THA)作为氨化试剂与3, 5-DNP的铵盐反应,得到ADNP。此过程常温反应2 d,后处理采用过硅胶色谱柱分离产物,收率50%。本研究以4-氯吡唑为原料,三甲基苯磺酰羟胺(MSH)为氨化试剂,通过反应得到ACDNP,并采用红外、核磁、质谱和元素分析手段进行表征,然后,对其中间体钾盐CDNPK的晶体结构和最终产物BCDNPA热性能也进行了研究。与文献相比,本方法明显缩短反应时间,常温反应20 h,简化了后处理过程,采用蒸馏水进行重结晶,收率提高至60.7%,通过对ANDP进行偶氮化处理,所得的BCDNPA产品热稳定性良好。
2 实验材料4-氯吡唑,常州市武进康达化工有限公司;浓硫酸,天津市化学试剂三厂;浓硝酸,天津市化学试剂三厂;乙醚,中国医药集团上海化学试剂公司;无水硫酸镁,分析纯,天津市化学试剂三厂;乙醇,分析纯,天津市化学试剂三厂;5%氢氧化钠的乙醇溶液,自制;无水乙腈,天津市化学试剂三厂;MSH试剂,自制。晶体结构用德国Bruker SMART APE II CCDⅡX-射线单晶衍射测定,元素分析、红外、质谱及核磁分别用德国Elementar公司Vario EL Ⅲ型自动微量有机元素分析仪、美国热电公司Nicolet 6700红外光谱仪(KBr压片)、美国瓦里安公司Varian 325 LC-MS液相色谱-质谱联用仪及德国Bruker AV II-400 MHz核磁共振波谱仪。
3 实验过程 3.1 合成和表征(1) 4-氯-3, 5-二硝基吡唑的合成

|
室温下,将15.9 g(0.16 mol)4-氯吡唑加入90.0 mL浓硫酸中,搅拌溶解,待用;冰水浴搅拌下,将43.2 mL浓硝酸滴加到90.0 mL浓硫酸,滴完搅拌30 min,控制体系温度在15~20℃,滴加上述配制的4-氯吡唑硫酸溶液,加完后缓慢升温至60 ℃反应2 h,将反应液倒入冰水中,搅拌至无色,乙醚萃取,无水硫酸镁干燥,蒸除溶剂、干燥得25.1 g白色固体,收率为84.1%。
1H NMR (DMCO-d6, 500 MHz),δ:8.76(s, NH);13C NMR(DMCO-d6, 125 MHz),δ:101.53, 150.39;IR(KBr, cm-1),υ:1 534, 1 421, 1 327, 685;元素分析C3HN4O4Cl(%):理论值:C 18.72, H 0.52, N 29.10;实测值:C 18.67, H 0.48, N 29.03。
(2) 1-氨基-4-氯-3, 5-二硝基吡唑(ACDNP)的合成
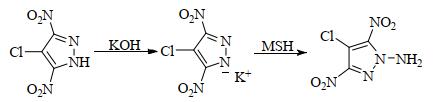
|
室温下,将2.0 g(10.4 mmol)4-氯-3, 5-二硝基吡唑溶解于20.0 mL乙醇中,冰水浴冷却至5~10 ℃,滴加11.6 g质量浓度为5%的氢氧化钾(10.4 mmol)乙醇溶液,室温反应0.5 h,过滤、乙醇洗、干燥得淡黄色固体4-氯-3, 5-二硝基吡唑钾盐;取0.93 g(4 mmol)上述钾盐加入30.0 mL无水乙腈中,滴加5.0 mL含1.29 g(6 mmol)MSH的乙腈溶液,室温反应20 h,过滤、蒸除溶剂、乙醇重结晶、干燥得0.62 g淡黄色固体,收率为74.7%。
1H NMR(DMSO-d6, 500 MHz),δ:7.90(s, 2H, NH2);13C NMR(DMSO-d6, 125 MHz),δ:132.27, 139.30, 144.27;IR(KBr, cm-1),υ:3 344, 3 267, 3 202, 1 621, 1 561, 1 500, 1 438, 1 332, 1 070, 997, 880, 768;元素分析C3H2N5O4Cl(%):理论值:C 17.36, H 0.97, N 33.75;实测值:C 17.28, H 1.05, N 33.66。
(3) 1, 1'-偶氮双(4-氯-3, 5-二硝基吡唑)(BCDNPA)的合成
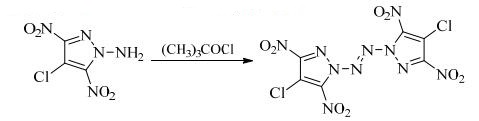
|
室温下,将0.34 g(1.64 mmol)ACDNP加入27.0 mL无水乙腈中,向反应瓶中持续通氮气,冰水浴冷却至0~5 ℃,加入0.54 g(5.03 mmol)次氯酸叔丁酯,自然升至室温反应20 h,减压蒸出乙腈,加入5.0 mL异丙醇搅拌,过滤、异丙醇淋洗、干燥得0.15 g淡黄色固体,收率为44.6%。
13C NMR(DMSO-d6, 125 MHz),δ:108.25, 132.25, 150.89;IR(KBr, cm-1),υ:1 569, 1 523, 1 425, 1 383, 1 318, 1 143, 1 059, 887, 816;元素分析C6N10O8Cl2(%):理论值:C 17.53, N 34.08;实测值:C 17.64, N 34.01。
3.2 CDNPK晶体的培育和结构测试取适量4-氯-3, 5-二硝基吡唑钾盐(CDNPK)溶解于无水乙腈中,室温静置,缓慢挥发10 d,得到淡黄色针状晶体。选用尺寸为0.37 mm × 0.31 mm × 0.26 mm的单晶,在X射线单晶衍射仪上,用经石墨单色器单色化的Mo Kα射线(λ = 0.071 073 nm)辐射,在296 (2) K下用ω/2θ方式扫描,在2.72°≤θ≤25.10°共收集到3 764个衍射点,其中独立衍射点1 412个(R(int) = 0.017 4)。所有计算由SHELXL97程序包解出,非氢原子坐标和各向异性温度因子经全矩阵最小二乘法修正,数据经Lp因子及经验吸收校正。晶体结构由直接法和Fourier合成法解出,经全矩阵最小二乘法对F2进行修正。最终偏差因子R1= 0.051 3,wR2= 0.177 5,GOF=1.092,精修参数为123个,最终差值在Fourier上最大残余峰为0.528×103 e·nm-3,最小残余峰为-0.952×103 e·nm-3。
4 结果与讨论 4.1 CDNPK的晶体结构分析化合物CDNPK属于单斜晶系,空间群C2/c,晶胞参数:a= 1.283 9(2) nm,b= 0.924 74(15) nm,c= 1.341 9(2) nm,α= 90°,β= 97.403(2)°,γ= 90°,V= 1.580 0(4) nm3,Z= 8,Dc= 1.535 g·cm-3,μ= 0.949 mm-1,F(000)=720。CDNPK晶体的结构及堆积图如图 1、2所示。部分键长和键角数据列于表 1。
|
|
表 1 CDNPK的部分键长和键角 Table 1 Typical bond lengths and angles of CDNPK |

|
图 1 CDNPK的分子结构 Fig.1 Molecular structure of CDNPK |

|
图 2 CDNPK的晶胞堆积图 Fig.2 3D packing of CDNPK |
由图 1和表 1实验结果可知,在CDNPK晶体结构中,每个最小不对称单元中含有一个中心K+和1个CDNP-。CDNP-在配位结构中起到螯合配体与桥联配体的作用,CDNP-结构中硝基基团的O1、O2、N3与K+分别构成螯合结构,结合堆积图,K+与不同CDNP-结构中硝基的N、O原子形成配位作用,形成了复杂的桥联结构,构成三维网状结构。
4.2 BCDNPA的DSC曲线DSC测试在敞开样品池、N2氛围条件下进行,流速30 mL·min-1,样品质量1.64 mg,升温速率10 ℃·min-1,温度范围30~500 ℃。测试结果如图 3所示。DSC曲线显示,在228~268 ℃有一个明显放热峰,该峰值温度253.88 ℃,为BCDNPA的受热分解放热峰。

|
图 3 BCDNPA的DSC曲线 Fig.3 DSC results of BCDNPA |
以4-氯吡唑为原料,经硝硫混酸处理后再与氢氧化钾反应生成CDNPK,并获得淡黄色针状CDNPK单晶,通过MSH试剂氨化合成了ACDNP,经偶氮化反应合成BCDNPA,并采用红外、核磁、质谱和元素分析手段对其晶体结构进行表征,其中间体CDNPK晶体结构也得到了深入研究。晶体结构测试结果表明,化合物CDNPK属于单斜晶系,空间群C2/c,a= 12.839(2) nm,b= 9.247 4(15) nm,c= 13.419(2) nm,α= 90°,β= 97.403(2)°,γ= 90°,V= 1 580.0(4)Å3,Z= 8,Dc= 1.535 g·cm-3,μ= 0.949 mm-1,F(000)=720。每个最小不对称单元中含有一个中心K+和1个CDNP-CDNP,有利于增强分子稳定性,降低感度。DSC测试结果表明,BCDNPA的热分解温度为253.88 ℃,说明该化合物热稳定性良好。
| [1] |
李亚南, 常佩, 王彬, 等. 氮杂芳环N-氨化物及其含能衍生物研究进展[J]. 高校化学工程学报, 2019, 33(2): 263-273. LI Y N, CHANG P, WANG B, et al. Review on N-amino compounds with N-heteroaromatic rings and their energetic derivatives[J]. Journal of Chemical Engineering of Chinese Universities, 2019, 33(2): 263-273. |
| [2] |
GAO H X, SHEREEVE J M. Azole-based energetic salts[J]. Chemical Reviews, 2011, 111(11): 7377-7436. DOI:10.1021/cr200039c |
| [3] |
ZHANG Q H, SHEREEVE J M. Energetic ionic liquids as explosives and propellant fuels:A new journey of ionic liquid chemistry[J]. Chemical Reviews, 2014, 114(20): 10527-10574. DOI:10.1021/cr500364t |
| [4] |
李亚南, 张生勇, 舒远杰, 等. 1, 1'-二硝氨基-5, 5'-偶氮双四唑钾盐的合成、结构及性能[J]. 火炸药学报, 2017, 40(4): 52-56. LI Y N, ZHANG S Y, SHU Y J, et al. Synthesis, structure and properties of dipotassium 1, 1'-dinitramino-5, 5'-azobitetrazolate[J]. Chinese Journal of Explosives & Prope-llants, 2017, 40(4): 52-56. DOI:10.14077/j.issn.1007-7812.2017.02.009 |
| [5] |
刘伟, 刘中星, 王建龙, 等. 基于加速量热仪的3, 4-二硝基吡唑绝热分解分析[J]. 科学技术与工程, 2018, 18(14): 121-125. LIU W, LIU Z X, WANG J L, et al. Adiabatic decomposition analyses of 3, 4-dinitropyrazole by accelerating rate calorimeter[J]. Science Technology and Engineering, 2018, 18(14): 121-125. DOI:10.3969/j.issn.1671-1815.2018.14.021 |
| [6] |
尹磊, 张至斌, 张建国, 等. 3, 4-二硝基吡唑的晶体结构[J]. 含能材料, 2016, 24(10): 965-968. YIN L, ZHANG Z B, ZHANG J G, et al. Crystal structure of 3, 4-dinitropyrazole[J]. Chinese Journal of Energetic Materials, 2016, 24(10): 965-968. |
| [7] |
SALAZAR L, ESPADA M, AVENDANO C. N-amination of 3-amino-1, 2, 4-triazole with hydroxylamine-O-sulfonic acid:Synthesis of 1, 5-diamino-1, 2, 4-triazole[J]. Journal of Heterocyclic Chemistry, 1990, 27: 1109-1110. DOI:10.1002/jhet.5570270455 |
| [8] |
RAJA D, PARITOSH R D, REDDY D, et a1. Synthesis of N-amino and N-nitramino-nitroimidazoles[J]. Terahedron letters, 2010, 51: 399-401. DOI:10.1016/j.tetlet.2009.11.046 |
| [9] |
HERVE G, ROUSSEL C, GRAINDORD H. Selective preparation of 3, 4, 5-trinitro-1 H-pyrazole:A stable all carbon-nitrated arene[J]. Angewandte Chemie International Edition, 2010, 49: 3177-3181. DOI:10.1002/anie.201000764 |
| [10] |
DALINGER I L, POPOVA G P, VATSADZE I A, et al. Synthesis of 3, 4, 5-trinitropyrazole[J]. Russian Chemical Bulletin, International Edition, 2009, 58(10): 2185-2187. DOI:10.1007/s11172-009-0301-2 |
| [11] |
DALINGER I L, VATSADZE I A, SHKINEVA T K. The specific reactivity of 3, 4, 5-trinitro-1 H-pyrazole[J]. Mendeleev Communications, 2010, 20(5): 253-254. DOI:10.1016/j.mencom.2010.09.003 |
| [12] |
RAVI P, GORE G M, VENKATESAN V, et al. Theoretical studies on the structure and detonation properties of amino-, methyl-, and nitro-substituted 3, 4, 5-trinitro-1H-pyrazoles[J]. Journal of Hazardous Materials, 2010, 183(1/2/3): 859-865. |
| [13] |
DUDDU R, DAVE P R, DAMAVARAPU R, et al. Synthesis of N-amino-and N-nitramino-nitroimidazoles[J]. Tetrahedron Letters, 2010, 51(2): 399-401. DOI:10.1016/j.tetlet.2009.11.046 |
| [14] |
YIN P, ZHANG Q H, ZHANG J H, et al. N-Trinitroethylamino functionalization of nitroimidazoles:A new strategy for high performance energetic materials[J]. Journal of Materials Chemistry A, 2013, 1: 7500-7510. DOI:10.1039/c3ta11356f |
| [15] |
ZHAO X X, QI C, ZHANG L B, et al. Amination of nitroazoles-A comparative study of structural and energetic properties[J]. Molecules, 2014, 19(1): 896-910. DOI:10.3390/molecules19010896 |